關于為什么Multiwfn算的出RESP電荷與Antechamber的有所差異
關于為什么Multiwfn算的出RESP電荷與Antechamber的有所差異
文/Sobereva@北京科音 2019-Sep-27
0 前言
RESP電荷的原理、細節以及計算在《RESP擬合靜電勢電荷的原理以及在Multiwfn中的計算》(http://www.shanxitv.org/441)有詳細說明,Multiwfn是計算RESP電荷最方便易用和靈活的程序。有Multiwfn用戶發現,Multiwfn算出的某些分子的RESP電荷與antechamber的不同,在此文我說一下原因。使用Multiwfn算RESP電荷的用戶不是很有必要看此文,Multiwfn給的結果肯定是合理的。但如果想了解一些細節的話也可以看看。
簡單來說導致差異的原因主要有兩點:
(1)擬合點的分布不同
(2)等價約束設置不同
下文涉及的體系的相關文件可以在這里下:http://www.shanxitv.org/attach/516/file.rar。本文用的是2019-Sep-26更新的Multiwfn 3.7(dev)版。
1 擬合點的分布不同帶來的差異
Multiwfn在計算RESP電荷的時候,對于3.6、3.7版默認用的是MK方式設置擬合點(以后版本有可能會改變,屆時看Multiwfn的RESP模塊界面上的選項提示便知默認的是什么),擬合點位置由Multiwfn自己的代碼設置,靜電勢由Multiwfn自己的代碼計算或者調用cubegen計算。Antechamber計算RESP電荷時是從Gaussian的pop=MK IOp(6/33=2) IOp(6/42=6)輸出文件里直接讀取MK擬合點的坐標和靜電勢。雖然MK原文雖然定義了擬合點的分布規則,即每個原子上在不同原子半徑倍數上有幾層擬合點,但在具體確定擬合點坐標上,不同程序的細節不同,比如在球面上的擬合點以什么算法生成排布?擬合點的密度是多少?在原子接縫的地方是否自動去除過密的擬合點?等等。由于Multiwfn設置的擬合點的位置和Gaussian在這方面有一些不同,導致Multiwfn和Antechamber算的RESP電荷會有些許差別,但這個因素造成的差異通常不大,也沒法說哪個程序更對,因為都是合理的。注:前述IOp(6/33=2)是讓Gaussian把擬合點的位置和靜電勢數值打印到輸出文件里的選項,而IOp(6/42=6)用來將擬合點的密度設為高于默認值。在Multiwfn的RESP電荷計算界面里也可以直接設擬合點的密度。
下面我們拿乙醇這個體系來對比一下。這里筆者直接用gview畫了一個乙醇,保存成了.mol2文件。然后用antechamber產生了對應的gjf文件,里面的關鍵詞是#HF/6-31G* SCF=tight Test Pop=MK iop(6/33=2) iop(6/42=6),原本帶著的opt我給刪了,對于本文的討論沒意義。然后用Gaussian 09 E.01進行計算,得到的chk文件我轉成了fch文件。
用Multiwfn載入fch文件,進入RESP計算界面,直接選標準兩步式RESP電荷計算,結果如下
Center Charge
1(C ) -0.250749
2(H ) 0.064449
3(H ) 0.064449
4(H ) 0.064449
5(C ) 0.547451
6(H ) -0.091298
7(H ) -0.091298
8(O ) -0.714589
9(H ) 0.407135
在計算第二步的時候提示用了以下等價性約束
Constraint 1: 2(H ) 3(H ) 4(H )
Constraint 2: 6(H ) 7(H )
再用antechamber計算RESP電荷,會出現一批文件,其中有兩個RESP計算模塊輸出的文件ANTECHAMBER_RESP1.OUT和ANTECHAMBER_RESP2.OUT。第一個就是第一步擬合的輸出,第二個就是第二步擬合的輸出,最終結果看第二個里面的下面的部分
Point Charges Before & After Optimization
no. At.no. q(init) q(opt) ivary d(rstr)/dq
1 6 -0.229693 -0.229799 0 0.003990
2 1 0.077688 0.059374 0 0.000000
3 1 0.077682 0.059374 2 0.000000
4 1 0.034340 0.059374 2 0.000000
5 6 0.464072 0.533889 0 0.001841
6 1 -0.057092 -0.086155 0 0.000000
7 1 -0.057098 -0.086155 6 0.000000
8 8 -0.720426 -0.720426 -99 0.001375
9 1 0.410526 0.410526 -99 0.000000
q(init)是第一步擬合之后產生的電荷,q(opt)是經過第二步擬合最終得到的RESP電荷。ivary是等價性約束關系,比如no.為3和4的原子的ivary是2,就代表2、3、4在擬合過程中保持等價,這和前面看到的Multiwfn自動用的約束完全一樣。8和9號原子的ivary為-99,代表它們在第二步擬合過程中電荷被約束為保持不變。
Antechamber這里產生的RESP電荷和前面給的Multiwfn算出來的有輕微差異,但頂多也就差0.01幾。如果我們想讓Multiwfn算出的電荷與Antechamber的精確一樣,那就在選擇開始計算RESP電荷之前先選一下選項8,然后再選1開始標準兩步式擬合,按屏幕提示的要求把Gaussian輸出文件路徑輸進去,這時Multiwfn就和Antechamber一樣從Gaussian輸出文件里讀取擬合點的位置和靜電勢了,輸出信息如下,和Antechamber給出的完全相同(個別原子仍有10的負六次方的差異是收斂限設置、舍入誤差等數值細節導致的,完全可忽略不計)
Center Charge
1(C ) -0.229798
2(H ) 0.059373
3(H ) 0.059373
4(H ) 0.059373
5(C ) 0.533887
6(H ) -0.086155
7(H ) -0.086155
8(O ) -0.720426
9(H ) 0.410526
2 等價約束設置不同帶的差異
下面我們看苯甲醛這個例子。哪怕Multiwfn也直接從Gaussian輸出文件里讀取擬合點的信息,結果與Antechamber給出的仍有輕微差異,這是由于兩個程序用的等價性約束設置不同所帶來的。
我們先用Multiwfn基于Gaussian里的擬合點信息計算RESP電荷。啟動Multiwfn,載入本文文件包里的C6H5CHO.fchk,進入RESP模塊,先選擇8,再選擇1,再輸入C6H5CHO.out的路徑,得到的結果如下
Center Charge
1(C ) -0.115868
2(C ) -0.111841
3(C ) -0.114944
4(C ) -0.146385
5(H ) 0.137991
6(H ) 0.134593
7(H ) 0.153718
8(H ) 0.139930
9(C ) 0.391423
10(H ) 0.059972
11(C ) -0.223448
12(H ) 0.155781
13(C ) 0.034578
14(O ) -0.495500
Sum of charges: 0.000000
RMSE: 0.001373 RRMSE: 0.079595
從屏幕上的提示可見,這個體系的RESP電荷計算過程只做了第一步,因為按照RESP電荷定義的規則,這個體系本身不需要做第二步。在第一步計算的時候沒有自動做任何等價性約束。
也用antechamber進行計算,從給出的ANTECHAMBER_RESP2.OUT中可以看到所有原子的ivary都為-99,即第二步相當于什么也沒做,因此最終得到的RESP電荷來自于第一步。然后看ANTECHAMBER_RESP1.OUT,輸出信息為
no. At.no. q(init) q(opt) ivary d(rstr)/dq
1 6 0.000000 -0.101223 0 0.003514
2 6 0.000000 -0.148518 0 0.002793
3 6 0.000000 -0.139285 0 0.002916
4 6 0.000000 -0.148518 2 0.002793
5 1 0.000000 0.136078 0 0.000000
6 1 0.000000 0.140390 0 0.000000
7 1 0.000000 0.147385 0 0.000000
8 1 0.000000 0.140390 6 0.000000
9 6 0.000000 0.404823 0 0.001199
10 1 0.000000 0.028701 0 0.000000
11 6 0.000000 -0.139285 3 0.002916
12 1 0.000000 0.147385 7 0.000000
13 6 0.000000 0.001412 0 0.004999
14 8 0.000000 -0.469734 0 0.001041
可見結果與Multiwfn產生的略有差異,特別是第11號原子。從ivary可見,第一步擬合的時候約束了4=2、8=6、11=3、12=7。此體系結構如下
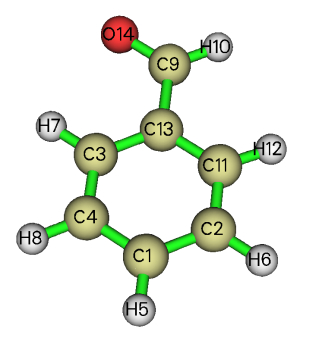
可見antechamber自動把苯環上左右對稱的原子電荷約束為了相同,而Multiwfn默認情況下沒有做這個約束,因此倆程序結果不同。
如果想讓Multiwfn也實現同樣的約束,自己寫個約束設置文件即可。寫個文本文件比如叫eqs.txt,內容為
4,2
8,6
11,3
12,7
在Multiwfn計算RESP電荷前,選擇設置等價性約束的選項,再選讀取約束文件,輸入eqs.txt的路徑,然后選擇2開始一步式RESP電荷擬合,此時自定義的約束會被利用上,結果為
Center Charge
1(C ) -0.101222
2(C ) -0.148519
3(C ) -0.139284
4(C ) -0.148519
5(H ) 0.136078
6(H ) 0.140390
7(H ) 0.147385
8(H ) 0.140390
9(C ) 0.404824
10(H ) 0.028701
11(C ) -0.139284
12(H ) 0.147385
13(C ) 0.001411
14(O ) -0.469734
Sum of charges: 0.000000
RMSE: 0.002020 RRMSE: 0.117100
可見此時結果和Antechamber給出的精確相同了。
對苯環上對稱的原子設置等價性約束并不是RESP電荷原文里提出的標準做法,純粹是Amber的開發者自己定義的規則。這個規則有一點道理,但帶來的壞處是會使得靜電勢重現性誤差增加。如上述信息所示,不做這個約束時RMSE是0.001373,做約束后就增加到了0.002020。對于Multiwfn用戶,這種約束想施加就施加,不想施加就不施加。如果懶得手動一條一條寫這種等價性約束設置,在Multiwfn中可以直接利用結構的對稱性自動產生等價性約束設置文件,看《RESP擬合靜電勢電荷的原理以及在Multiwfn中的計算》(http://www.shanxitv.org/441)第3.5節的例子。