首個完全基于GPU的量化軟件-TeraChem雜談及真實性能測試
2019-Mar-3注:在文末補充了全新的測試,以反映目前主流硬件下的情況
首個完全基于GPU的量化軟件-TeraChem雜談及真實性能測試
文/Sobereva 2012-Apr-30
TeraChem是第一個公布于世的、完全基于GPU運算的從頭算量化程序,2010年5月發布第一版。雖然很早就聽說過此程序,但直到近日才終于有機會親自把玩,此文將雜談此程序的一些特征,并且實際測試一下此程序的性能,看看到底有沒有官方吹得那么玄,是否值得為此投資。本文的測試是確保公平的,鑒于寡人知識水平不敢說在討論上一定正確,但可以保證沒有任何偏向。
1 簡介
TeraChem是2011年剛評上美國科學院院士的Todd Martinez和他的一個俄羅斯博士生Ufimtsev基于CUDA開發的充分利用GPU運算能力號稱使計算速度有飛躍式提升的從頭算量化程序,算法基礎是他們在JCTC上于2008、2009年發的三篇文章(JCTC,4,222、JCTC,5,1004、JCTC,5,2619),分別闡述了利用GPU進行雙電子積分、直接SCF、解析梯度的實現方法。雖然也有其它一些程序號稱支持GPU加速,比如Q-Chem、Firefly、GAMESS-US等,包括Gaussian也高調宣布與nVidia合作將在未來支持GPU加速,但是它們基本上都是在原有的基于CPU的代碼、算法基礎上去改造而支持GPU加速,而TeraChem則是第一個完全從頭寫的,根本目的就是充分發揮GPU運算能力的量化程序。TeraChem要求至少有一個支持CUDA的GPU方可運行。由于目前只有nVidia的GPU支持CUDA,AMD顯卡用戶就別指望了。
TeraChem現在屬于PetaChem公司的產品,也是此公司旗下目前唯一的產品,根據購買量價格從315到900刀不等。在中國由坤成基業公司代理,但價格不明朗,貌似此公司極力想將這款軟件連同他們的帶Tesla卡的服務器捆綁銷售。
此程序雖然還遠遠不夠成熟,功能還很有限,撰文之時此程序最新版本1.5版剛剛發布,直到這個版本才剛剛支持了d基函數(也有說法是1.45版就已經支持了),某種意義上才算剛摘掉了"Toy"的帽子。此程序主要能做的事就是算單點能、幾何優化、找過渡態、BOMD、算偶極矩和極化率(超極化率算不了)。支持的理論方法就是HF、GGA/雜化DFT,R/U/RO形式都支持。嚴重的局限性是尚不支持激發態計算,頻率也算不了(起碼給個數值Hessian嘛)、不支持任何后HF方法、不支持對稱性。隱式溶劑模型、算NMR、BSSE、贗勢、外加電場等等零零碎碎的功能更是一概沒有。雖說支持QM/MM,但MM區域只能是TIP3P水,因此實用意義很有限,Amber12倒是支持掛著TeraChem做QM/MM。此程序直接支持Grimme的DFT-D2/D3,直接附帶了NBO6.0,這倒是挺有眼光,不過NBO6.0分析時還很容易崩潰,而且一些NBO功能不能用,比如NBO deletion(奇怪的是,筆者撰文時NBO的官方最新版本仍然只是5.9,雖然早已聽聞6.0的消息,但不明白為何6.0先出現在了TeraChem程序包里)。TeraChem使用的基函數就是一般分子量化軟件用的高斯型基函數。
此程序比較新,而它的license是和網卡MAC綁定的,也沒有個能夠公開下載的體驗版,這就造成了此程序很難讓廣大量子化學研究者有機會接觸到它,我想,愿意去花不少錢購買一個很稚嫩又很未知的東西的人不會很多。好像這程序剛開始推廣那陣子可以向開發者免費申請試用,但現在已沒這個好事了。雖說前些日子在北師大搞的TeraChem培訓班倒是讓一些人有機會使用,但是產生的影響力我想還是很有限的。據悉可以向和TeraChem交情很深的北師大的于建國教授申請索要兩個月的demo license,有興趣者可以聯系他(PS:過期之后改掉系統日期就能繼續用)。
2 使用感受雜談
此程序文件結構很簡單,包括一個300MB出頭的可執行文件、NBO6.0可執行文件、license文件、userguide的pdf文檔、基組庫文件夾,還有幾個零碎的沒什么用的文件。基組庫文件夾里每種基組對應一個文件,比如6-31+G*基組對應于6-31+gs文件,內容和EMSL基組數據庫里的數據是直接對應的。雖然也帶了含有高角動量函數的基組,比如cc-pvqz,但由于此程序尚不能支持f及以上角動量,所以這些基組目前也根本沒法用。
此程序手冊編寫得比較簡單,才32頁而已,只有比較成熟的量化軟件手冊厚度的1/10的級別(寡人寫的Multiwfn的手冊厚度都是它的5倍咧)。雖然手冊編寫得這么省事據說是開發者沒時間所致,但我想,費很大力氣寫了很多代碼,但是沒個詳細描述其功能的手冊,用戶不會用,那么等于白費力氣。好在TeraChem使用起來比較容易,有一定經驗的量化工作者稍微摸索一下就會基本使用,但是有些該細說的比如球形邊界條件卻說得太抽象含糊。
TeraChem 1.5只有Linux版,建議用RHEL5.5及以上版本,要求安裝CUDA驅動和CUDA 4.0 Toolkit。貌似不提供源代碼包而只提供編譯好的文件。只需要將TeraChem環境變量指向license文件就能直接用了,比如export TeraChem=/sob/TeraChem/license.dat。運行時輸入比如./TeraChem [輸入文件名] 就行了,如果不重定向到文件中的話運算過程的信息會輸出在屏幕上,各類結果信息比如幾何結構、Mulliken電荷會輸出在scr目錄下的不同文件中。如果涉及到NBO分析的話,需要設定NBO6.0可執行文件路徑的環境變量,例如export NBOEXE=/sob/TeraChem/nbo6.exe。
輸入文件的編寫比較容易,比如一般的計算任務(這里是結構優化)只需要如下這么寫,含義不言自明,前后順序隨意。如果要加注釋就寫在#號后面。
basis 6-31gss
charge 0
spinmult 1
method wb97x
run minimize
coordinates MN-NN.xyz
end
比如像加上DFT-D3色散校正,加一句dftd d3就行了。手冊可以在其網頁上免費下載http://petachem.com/pricing.html,關鍵詞并不多,有興趣者不妨看看。目前直接支持的結構文件格式是xyz和pdb。
于建國教授和幾個學生開發了一個TeraChem的前端圖形界面SimuTera(收費),可以構建分子、創建輸入文件、本地/遠程提交、觀察結果,有點類似Gaussview和Gaussian的關系。TeraChem官方主要是推薦用VMD觀看生成的結構、動力學軌跡和分子軌道。TeraChem與VMD結合可以很方便地實現Interactive molecular dynamics(IMD),也就是在TeraChem輸入文件中寫個上比如imd 54332,那么TeraChem計算開始前會等待VMD連接,在VMD里進入IMD Connect插件并且填上主機IP和54332這個端口號并點連接,動力學計算就會開始,每一步的結構變化都會實時傳到VMD的圖形界面上。結構優化、過渡態搜索過程也都支持這種方式觀看。據說以后還能像NAMD那樣,支持交互式施加外力操作,也就是在VMD圖形窗口中通過在某處拖動鼠標來直接給相應區域原子施加特定方向和大小的力。
雖然此程序的幾何優化還是常見的算法,包括L-BFGS、共軛梯度、最陡下降法,但是,搜索過渡態居然只支持NEB,這點值得稍微吐槽。NEB方法不常出現在分子量子化學軟件中,而常用于第一性原理的軟件中,我曾經在《過渡態、反應路徑的計算方法及相關問題》(http://www.shanxitv.org/44)當中介紹過其原理。NEB方法必須提供過渡態前后兩個位置的初猜坐標(這一點類似于QST2),這顯然比起常用的基于GDIIS和準牛頓法極其變種那樣只需要提供一個過渡態初猜坐標的方法麻煩很多,雖說額外的好處是能順便得到這兩點間的能量極小路徑(如果提供的兩個坐標就是過渡態連接的兩個能量極小點,就得到了IRC)。限于時間,筆者沒具體測試這個功能。我認為,在TeraChem中加入一種只需要單個初猜結構的搜索過渡態的功能應該不是什么難事,而且也應該是當務之急。
TeraChem的從頭算動力學是主要賣點之一,由于號稱運算速度快,可以做較大體系較長時間的動力學。在這方面一個有趣的功能是每隔幾幀輸出一次分子軌道信息,通過VMD可以看到分子軌道圖形在模擬過程中的動態變化過程。雖然這看起來很有趣,在視覺上挺吸引人,但是我倒覺得沒太大實際意義。TeraChem的動力學的設定還不算很完善,雖然支持控溫,但是沒法指定溫度變化過程,沒法方便地由用戶設定初始速度,沒法加限制勢或拉伸力。此程序只支持比較怪異的球形周期邊界條件,竟不支持一般的矩形盒子邊界條件。
量化程序之所以沒有像分子動力學程序那樣那么早就支持GPU加速并付諸于實際應用,一方面是量化涉及到的算法不僅種類多而且過程復雜得多,另一方面是早年間的GPU不支持雙精度浮點,這難以滿足量化計算數值方法的精度要求(盡管單精度浮點也不是完全在量化中沒用,比如專門有人提出基于單精度浮點的MP2算法)。直至nVidia的G200系列GPU開始(即民用的從GTX260開始,Tesla系列從C1060開始)才支持雙精度浮點,這才使GPU在量化上的普遍應用成為了可能。因此我起初以為TeraChem是完全的雙精度,看了手冊才知道原來這程序在內部默認是單雙精度混合(這有點像amber的CUDA版的處理),對于數值大小高于閾值的雙電子積分以及涉及到數值累加的變量一律用雙精度以保證精確度,而數值較小的雙電子積分采用單精度。這種混合方法是考慮到目前nVidia家用的Geforce系列GPU的雙精度運算能力只有單精度的幾分之一,而Tesla和Quadro系列的雙精度性能也只有單精度的一半。
我個人比較希望此程序能輸出.wfn文件或者Molden輸入文件,由于號稱此程序算大體系比較容易,這樣Multiwfn也就能分析很大體系波函數信息了。也許有空的話我也會嘗試想辦法將TeraChem的輸出信息轉換成wfn格式。
手冊中說不推薦用GTX580和Tesla C2075運行此程序,這挺麻煩,而現在基于新架構Kepler的GTX680、GT640/630已經推出了,恐怕TeraChem在其上運行會更不如意。TeraChem需要盡快著手支持新硬件。硬件更新換代帶來的問題在很多為GPU深度優化的基于CUDA的產品中都存在,amber也是,記得其開發者說當前amber的CUDA版為Fermi架構做了很多優化,直接上GTX680可能會有些問題。
值得一提的是,雖說此程序基本上完全基于GPU,但實際上GPU在運行時CPU仍然有不低的占用率,可以算的尺度也受主機內存大小的限制。
3 性能測試
3.1 測試的意義和測試環境
TeraChem官方宣稱這軟件能帶來比基于CPU的程序幾十倍的性能提升。實際上,每款支持GPU計算的軟件都愛這么宣揚,少則說提升7、8倍,一般都說提升幾十倍,高則乃至上百倍。每次nVidia的人做報告的時候也是這么使勁吹,好幾年來我耳朵聽得已經起繭子了。2011年《物理化學學報》27卷2019頁的《GPU引發的計算化學革命》也是有很多類似的說法,列了不少數據,都說提升了很多倍。然而我并不喜歡這類含糊的說辭,有大躍進時代鼓吹畝產萬斤之嫌。對待GPU加速切不可頭腦過熱,我們必須明確這驚人的加速比到底來自于什么GPU和什么CPU在比較,究竟這看似引發“革命”的加速比到底靠不靠譜、有無意義,謹慎分析究竟我們要付出多大代價才能享受到這收益,是否能真正給研究工作帶來實實在在的效益。比如我用過的一些GPU加速視頻轉碼的軟件得到的畫質很差,等于GPU加速根本沒帶來任何用處;而很多宣稱支持GPU加速的計算化學軟件只在用處不大的功能上性能提升巨大,比如說分子動力學軟件在GB溶劑模型下加速比很大,也是每次都被拿來在臺面上鼓吹的數據,但最常用的顯式溶劑下加速比則明顯沒那么高,資金投入上未必選擇GPU代替CPU更劃算,可以參看以前在我寫過的一篇《小測Tesla C2050在amber11上的GPU加速性能》(http://www.shanxitv.org/67,由于硬件價格的變動文中一些分析可能不再合適了)。我從數年前nVidia剛推廣CUDA時就看好GPU加速在計算化學上起到的巨大作用,也不否認“革命”這種說法,但是在現階段一定要理智。說了這么多主要是想表明為什么我要對TeraChem做真實、客觀的性能測試,因為大多數軟件的官方數據和說辭歷來是不可盲目信從的,往往刻意掩蓋重要信息或設定不公平的參數而讓自己的成績遠高于對手,報喜不報憂,尤其是打著“GPU加速”這樣相對較新概念的旗號時更容易讓用戶相信一些夸張的宣傳。
我們將考察TeraChem計算單點能時隨體系尺寸增加計算時間的變化、考察不同配置下性能的差異、考察計算精度,并與最常用、最成熟的量化軟件Gaussian的03版相比較看看到底能TeraChem有多大的性能優勢。實際上不同量化軟件間性能對比測試是很不容易進行的,雖然基本算法都一樣,但在具體處理上,比如如何判定已經收斂、用什么樣的加速收斂方法、用什么方式加速積分、如何構建初猜等等都是不同的。但我想,作為用戶,我們主要關心的是對同一個體系不同軟件到底花上多少時間能得到精度差不多的結果。所以我對默認設置不做多余的調整,因為用戶一般也并不去調。TeraChem是通過DIIS Error來決定收斂的,這和Gaussian用的能量及密度矩陣變化判據不同因此沒法完全一致,但由于G03用戶計算單點能時一般就用默認判據,而且G03在默認判據下收斂時DIIS誤差通常和TeraChem默認的收斂限0.00003差不多,所以我認為兩款程序在默認收斂判據下的耗時是有可比性的。另外,TeraChem默認的DFT數值積分格點精度偏低,這直接影響結果精度和耗時,所以輸入文件里用了dftgrid 2,和G03默認的基本在一個級別,即每個原子用六七千個點。TeraChem的數據是程序自身統計出的實際消耗時間,由于運算前還有初始化GPU設備過程,因此實際用時比下文給出的要多出數秒鐘。Gaussian的運行時間是Linux的time命令給出的時間。本文所謂時間都是指wall clock time而非CPU時間。若未注明,TeraChem用的一律是默認的動態單雙精度混合模式。
測試涉及到C2050和GTX480的結果都是在第三方的帶有兩塊C2050+兩塊GTX480的雙路E5620+24GB的機子獲得的結果,估計用的是Intel 5520芯片組,這四張卡各自的PCI-E 2.0口的具體分配不清楚,當然若是4*8x則是最均衡的。涉及到i7-2630QM和GTX460M(1.5GB顯存)的結果都是在寡人自己的Toshiba X500-01R筆記本電腦上測的結果(8GB內存,RHEL6U1系統)。TeraChem為1.5版,Gaussian03為E.01。雖然i7-2630QM支持超線程,但對于本文的測試任務在Gaussian下只用4線程更快,所以測試都是nproc=4下進行。
3.2 計算速度測試
筆者先用一系列下面這種鏈狀聚苯分子進行測試,后文中的N代表分子中苯環的數目。總原子數是2+10*N。皆C1對稱性。計算級別使用對于大體系的經典搭配B3LYP/6-31G*。
測試結果如下
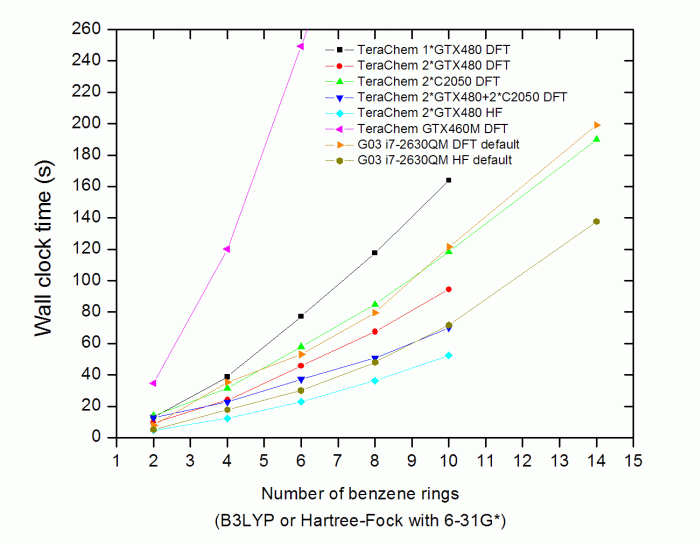
GTX460M在N=8、N=10、N=14時的耗時在圖中未顯示,分別為387.1s、547.4s、901.3s。
從這個圖中,我們可以得到這些結論:
(1)兩塊GTX480比起一塊GTX480的時候性能提升很多,當原子數多時,提升將近翻倍。兩塊C2050的性能竟然不如兩塊GTX480,這是有些出乎意料的,按理說前者的雙精度浮點性能比后者強得多。但是,這個結果也在情理之中,因為GTX480的核心及顯存頻率都比C2050高一些,而且前者的流處理器是480個而后者是448個,因此單精度性能顯然GTX480更強。已經提到,TeraChem是單雙精度混合運算,所以GTX480在此程序中表現占優是可以理解的。而兩塊GTX480和兩塊C2050一起用時并沒比兩塊GTX480時強太多,不知是此程序的問題,還是PCI-E帶寬或其它什么原因造成了瓶頸。考慮到C2050比GTX480貴好幾倍,從性能上看使用TeraChem完全不值得買Tesla產品,用高端的Geforce系列GPU最劃算。
(2)TeraChem算大一些的體系的時候還是很耗顯存的,在計算N=14的聚苯時擁有1.5GB的GTX480已經out of memory了,無論是單塊還是兩塊,或者和C2050混用時都因此報錯而無法進行。所以,Tesla產品在TeraChem上相對于Geforce系列產品來說的優勢也就是能計算更大的體系(例如C2050和C2070分別為3GB和6GB顯存)。然而很多非公版的Geforce產品的顯存都比公版標配的大,通常大一倍,但價格仍比較合理,因此我建議選擇這些非公版大顯存的高端Geforce卡用于TeraChem計算。奇怪的是,盡管寡人的GTX460M也是1.5GB顯存,但計算N=14的聚苯時很正常。
(3)GPU的性能對運算速度影響明顯。GTX460M只有192個流處理器,為GTX480的40%,而且顯存位寬也只是其一半,頻率也更低,因此,運算速度只有GTX480的不到1/3。
(4)TeraChem的DFT比HF慢的程度相對于G03來說略大一些。
(5)TeraChem和G03都在N=2至N=14的范疇內顯示出線性標度的特征,曲線幾乎是直的。雖說這兩個程序都沒有納入稱得上線性標度的算法,但其實這結果也并不意外,因為線性分子通過簡單的積分截斷就很容易展現出準線性標度。而實際上很多人專門搞出一些號稱線性標度的方法用在日常遇到的非線性體系上效果很不理想。所以,對于日常研究的各種體系,這兩款程序一般不會隨著原子數的增多而曲線保持這么平坦。
(6)最后是最關鍵的一點,也就是,TeraChem的計算速度遠遠沒有它鼓吹得那么快!!!從圖可見,i7-2630QM這種折合于普通臺式機1000塊錢出頭的CPU在G03上的運算耗時基本上和2*C2050在TeraChem上的運算速度持平,哪來的號稱的神乎其神的幾十倍的提升?可以估算出現在主流的雙路計算服務器下G03的運算耗時差不多能和2*GTX480+2*C2050耗時持平。而且,如果GPU不是那么高端,只是主流家用級別的話(GTX460M折合于GTS450這檔次),那么還遠遠不如一個普通四核CPU在G03上算得快。
TeraChem允許自行設定使用完全單精度、完全雙精度、還是動態/靜態單雙精度混合運算。這里對8個苯環B3LYP/6-31G*的情況比較了默認情況、純單精度和純雙精度的計算耗時。
2*GTX480 單精度:54.3s
2*GTX480 雙精度:205.9s
2*GTX480 默認:67.5s
2*C2050 單精度:70s
2*C2050 雙精度:272.1s
2*C2050 默認:84.9s
很明顯,純雙精度時速度比單精度時慢得多得多,而動態混合單雙精度并不比單精度多耗時多少。有點令我奇怪的是,明明C2050在雙精度浮點性能上高于GTX480很多,但是從實測數據上看,依然像純單精度浮點時一樣吃虧。
前面我們只測了線性分子,這有點太理想化了,所以下面我們也測一個更實際的分子,咪唑配位的鎂卟啉,看看是否也有相似的結論。其化學組成為H18C24N6Mg1,如下圖所示
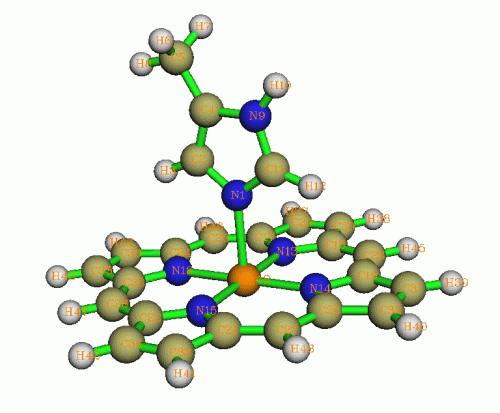
計算方法仍是B3LYP/6-31G*
1*GTX480 105.6s
2*GTX480 62.2s
1*C2050 137.5s
2*C2050 80.3s
2*GTX480+C2050 50.8s
GTX460M 348.8s
G03 i7-2630QM 156.5s
幾種GPU的運行耗時和前面結論基本一致。這回終于TeraChem比G03占了上風,但是,一個GTX480跑TeraChem的耗時僅僅比i7-2630QM跑G03的耗時降低了三分之一而已,遠遠沒有傳說中的夸張。對于擁有對稱性的體系,尤其是高對稱性體系,TeraChem的優勢必然還會喪失掉。GTX460M+TeraChem的效率依然明顯不如i7-2630QM+G03。
3.3 計算精度測試
對于咪唑配位的鎂卟啉,下面比較一下TeraChem不同模式下的計算結果的精度,并與可靠性受到公認的Gaussian的結果進行對比。不同量化程序間Hartree-Fock的結果比起DFT的更有可比性。
Gaussian03 默認 -1445.7243754
Gaussian03 SCF=Tight -1445.7245352
TeraChem 默認 -1445.7245359061
TeraChem 單精度 -1445.7247432920
TeraChem 雙精度 -1445.7245309871
可以看到,TeraChem默認的動態單雙精度混合情況下和純雙精度時結果很接近,用原子單位時差異僅在小數點后第六位,在一般研究中其差異可忽略。而單精度的結果偏離雙精度結果稍大一些,但也僅在小數點后第四位有偏離。鑒于TeraChem默認精度下與純雙精度結果符合很好而耗時又不比純單精度時增加很多,建議一般計算時都用默認精度。TeraChem的結果和Gaussian在SCF=Tight收斂標準時符合得很好,說明TeraChem這款較新的程序的結果可信度是有保證的。G03在默認收斂標準下的結果和Tight標準下的稍有偏離,不過也僅折合于相差0.42KJ/mol罷了,除非很高精度的計算,這點誤差可以接受。從這個體系看,TeraChem在默認標準下比G03默認標準下精度高一點點,但G03再多做兩三輪迭代也是能達到相同精度的,不會影響上文對這兩款程序計算效率的討論。
DFT的交換相關泛函在量化程序中都是用格點方式近似地數值積分,不像HF的結果那樣能得到精確值,而不同程序在DFT的積分格點設定上差異經常比較大,所以不好比。但這里仍然嘗試比較一下兩款程序在B3LYP/6-31G*下N=14的聚苯的計算精度。
Gaussian03 默認-3235.9677616
Gaussian03 SCF=Tight -3235.9678984
TeraChem 默認 -3235.9678361702
可見TeraChem默認精度下的結果依然與G03在Tight收斂標準下的結果相符得很好,且依然比G03在默認收斂標準下結果精度稍高。
3.4 關于TeraChem官方測試數據
下圖是直接從TeraChem官方網站上復制來的官方測試數據
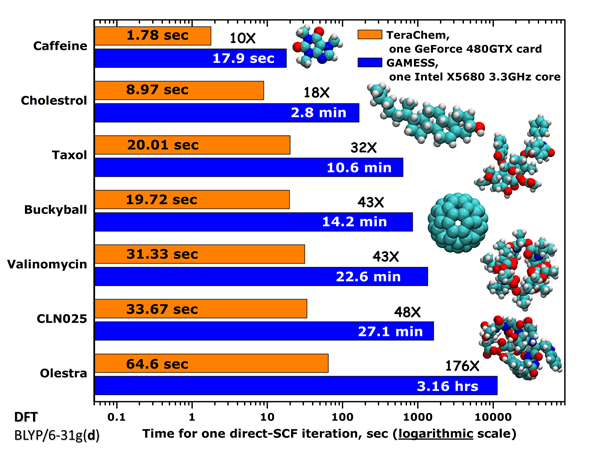
乍一看這圖,感覺TeraChem簡直神了。然而,本文的實測數據卻顯示TeraChem的計算效率還是和最主流的量化程序G03在一個數量級,完全不像官方吹噓的那樣神,難道官方在虛假宣傳?
這里我們把注意力放在C60的測試數據上,因為大家都很容易構建其輸入文件并在自己的機子上實際測試。官方是在BLYP/6-31G*下計算的,圖中是SCF每一輪迭代的用時。官方給的GAMESS測試數據慢得離譜,在比較高端的XEON X5680上(六核,3.33G),居然一輪迭代要花上14.2分鐘,十幾輪迭代收斂后,相當于要花上兩三個小時!這數據明顯有問題。倒是GTX480每輪迭代花19.7s憑本人感覺確實屬實(筆者沒來及實測)。從圖例可見,GAMESS寫的是"one Intel X5680 3.3GHz core",究竟這是指一個X5680 CPU還是指它的一個核心的速度?為了搞明白,我在i7-2630QM上四核并行測了一下GAMESS-US目前最新的2011-AUG-11_R1版,每輪迭代僅耗時202s(未考慮對稱性),若是X5680六核并行,那么折算起來也就是100秒出頭而已,只能說單GTX480+TeraChem是X5680+GAMESS-US效率的5倍,而根本不是圖中寫的43倍。而官方數據,很明顯只是用X5680的一個核來和GTX480的480個流處理器來對比,這還有公平可言么!?即便是一個核,官方給的GAMESS-US的耗時也比我估算的長不少,是什么原因這就不好說了,也許是編譯的問題。
對這個體系,使用i7-2630QM在Gaussian03中每輪迭代只花了18秒,這比起單GTX480在TeraChem中甚至還略快一點,而且這還是未考慮對稱性的情況下的耗時。因此,TeraChem實際效率只能說和當前主流量子化學程序平級,根本不能指望通過使用TeraChem能夠令計算耗時有質的降低,官方的圖表根本沒現實意義。我不排除TeraChem在研究很大體系的問題上可能比Gaussian會有明顯一些的速度優勢的可能性,然而,TeraChem對顯存容量要求比較高,Geforce系列顯卡能研究的體系尺度還是比較有限的。而使用顯存高達6GB的一些Tesla產品雖然足夠研究頗大的體系,但是過高的價格導致這投資明顯不如用在CPU上更劃算。
幾何優化也是十分重要的功能值得測試,但限于時間和篇幅本文只能對計算單點能進行測試。在幾何優化算法和穩定性上TeraChem肯定遜于Gaussian(于建國教授也提到過這點。而且Gaussian的主要開發者之一Schlegel是幾何優化算法方面的頂尖專家)。
4 結論與評價
雖然首個完全基于GPU的TeraChem在宣傳數據上看似效率高得十分驚人,并成為此程序的最主要賣點,然而經過本文的實際測試,發現其效率遠遠被夸大了,在日常研究工作中很難帶來顯著的收益,而且此程序還比較稚嫩,還很不完善。雖然說TeraChem不值得購買可能會過于傷害一部分人的感情,并可能由于本人對此程序了解不夠深而冤枉了此程序,但至少我要說,購買TeraChem需謹慎,切不可盲目信從其官方宣傳,想買的話應當實際測一下它在你想研究的問題上的性能(坤成基業網站上可以申請機時免費測試此程序),并根據GPU和CPU產品的硬件價格理性分析一下是否值得投資。
我對TeraChem程序本身不抱有任何偏見,但是不得不說現階段TeraChem的性能表現令我失望,遠低于我最初的預期,若不是我仔細地對比一下它和Gaussian的性能,沒準我就被它的夸張的宣傳所迷惑了,實際上Gaussian的運算速度相對于其它很多量化程序也并不算高。那么,既然TeraChem沒帶來多大實際效益,它在現階段究竟帶來了什么?我想,TeraChem目前的存在意義或許只是很好地證明了基于GPU的量子化學程序能干和傳統基于CPU的量子化學程序相同的工作,并且結果符合得很好。我希望TeraChem能踏踏實實地發展下去,努力提高效率(包括改進涉及GPU的代碼,也包括引入RI近似、FMM等算法),完善功能,不要為了急于提高銷量而在性能上放衛星。我期待著GPU的運算能力終有一天能使廣大量化工作者切實獲益,使量子化學問題的計算速度有普遍性的飛躍。而作為用戶,當前切不可被各類GPU計算程序鋪天蓋地熱火朝天的宣傳(TeraChem就是典型例子)搞得頭腦過熱,否則只會花冤枉錢、浪費時間。
還要指出的是,基于GPU和基于CPU的程序的性能對比必須明確到底是什么硬件型號之間在對比,光是說諸如“XXX算法利用GPU加速技術使計算速度提升了20倍”這是沒有任何意義的。如果是Geforce 205這種只有8個低頻流處理器的最低端的支持CUDA的GPU產品去和XEON E7-2870這種頂級的10核CPU相比,說GPU比CPU快顯然是天方夜譚。在很多基于GPU的程序上,時下中檔GPU表現的效率也就是和中高檔CPU平級,而在TeraChem上更甚,若沒有一塊高檔GPU,就更別指望此程序能有多少用處。
2019-Mar-3的補充:
有些人嫌本文內容已經過時了,認為GPU性能后來又大有發展。這里筆者從Terachem官網上取了一個比較新一些的測試任務,是Terachem v1.93P結合GTX1080Ti在BLYP/6-31G*下跑一個110原子的有機分子的能量+受力,耗時是48s。筆者用2*E5 2696v3的機子(詳見《淘寶店購買雙路2696v3服務器的過程、使用感受和雜談》http://bbs.keinsci.com/thread-6310-1-1.html)用ORCA 4.1.1在RI-BLYP結合與6-31G*差不多大的基組def2-SV(P),對這個體系也進行能量+受力計算,花了61s,由于倆程序的積分格點精度、收斂限等設定存在差異,可以認為耗時沒有區別。目前一個主流6核PC機+1080Ti得12000,Terachem程序一臺機子授權費就得大約10000塊錢,總共兩萬多,而目前與2*E5 2696v3性能持平的機子也才兩萬(如果用ES版CPU,那總共一萬多點就能拿下來),ORCA還是免費的,可見即便在2019年3月的時候,Terachem相對于基于CPU的量化程序在性價比上依然沒有任何優勢。
另外,雖然Terachem后來又做了改進,但還是功能局限性極大,諸如還是不支持d以上角動量基函數,連解析Hessian都沒有,明尼蘇達系列泛函(如主流的M06-2X)一個都不支持,從功能上看Terachem幾乎毫無使用價值。