Gaussian型Cube格點文件分析、運算程序GsGrid V1.6
注意:GsGrid的全部功能早已經融合到了Multiwfn (http://www.shanxitv.org/multiwfn)當中作為主功能13,故GsGrid不再更新。
GsGrid V1.6
Extract data from Gaussian grid file and grid file calculation
Programmed by Sobereva, 2009-Oct-7
Bug report or recommend: Sobereva@sina.com
1. 更新記錄:
V1.0 最初版本,包含前四項功能。
V1.1 將輸入輸出單位由以前的波爾半徑改為埃。加入了輸出一定范圍內平面的平均值的功能。
V1.3 修正了以前版本嚴重bug(即提取XZ平面數據得到的結果有時會偏差較大),新增功能8、功能9,用于得到自定義平面的格點數據。
V1.4 提高了output.txt中的數據輸出精度(坐標精度不變),小數部分擴展為15位(整數部分最大5位),加入了格點文件計算功能。
V1.5 修正格點文件類型判斷方式,使cube關鍵字生成的分子軌道格點文件可直接被gsgrid正確讀取。新增支持多分子軌道格點文件。
V1.5.1 修正一個載入某些格點文件時出錯的bug。
V1.5.2 修正讀取多MO的格點文件失敗的bug。
V1.6 加入了提取等值面上格點的功能,并可以將另外格點文件投影到某等值面上
2. GsGrid簡介
Gaussian
提供了cubegen工具,以及cube、prop選項可以得到格點文件。缺點是數據點沒有對應的坐標信息,而且也無法提取某個平面的數據作圖。雖然支持
格點文件的可視化軟件很多,GaussView較新版本及ChemCraft可以直接顯示某個自定義平面上的等值線(前者支持)或填色圖(后者支持),但
是程序自建繪圖功能極為有限,較為粗糙,不適合作為文獻插圖,無法導出數據在專業作圖軟件中自定義繪制,另外這兩個軟件也是收費的。至于molden,沒
有純粹的windows版本,界面頗不友好。GsGrid主要為解決這些問題制作。
對于格點運算,雖然ChemCraft和cubman都能實現,但是所支持的運算操作各有不足,gsgrid從v1.4開始支持豐富的格點運算操作,范圍超過ChemCraft和cubman支持操作的并集。
雖 然很多軟件,如果gaussview、Moliso等都可以將某個屬性投影到某個等值面上并以顏色表示數值大小,比如最常見的就是將分子靜電勢(MEP) 投影到電子密度為0.001的等值面,用以研究分子VDW表面的靜電勢。然而這些軟件并不提供將等值面上的數值輸出的功能,只能用顏色顯示,GsGrid 彌補了這一不足。
2. GsGrid功能介紹
GsGrid目前支持6大類功能
第一類功能(程序中第1項):把格點文件的數據提取出來,附上每個數據點的坐標。見例1。
第二類功能(程序中第2-4項):選取指定的XY/YZ/XZ平面,得到這個平面上的數據點以便做圖。見例2。
第三類功能(程序中第5-7項):將一定范圍內平面的數據取平均輸出,比如Z值在-3至3.5埃之間的平面的數據取平均輸出。
第
四類功能(程序中第8-9項):通過指定三個原子,或者自行輸入三個點的坐標,定義一個平面,得到此平面的上的格點數據。此平面上的數據點可以直接輸出,
但是數據點并不在一個平面上,不方便做圖。程序可以將此平面與XY平面的相交線做為軸,旋轉此平面,使數據轉移至XY平面,即此時數據點Z值皆為0,隨后
可通過繪圖軟件做contour圖。見例3。
第五類功能(程序中第10項):
格點文件中每個數據對某個常數進行加減乘除(子菜單1/3/5/7項)
格點文件中每個數據對另一個格點文件對應項進行加減乘除(子菜單2/4/6/8項)
格點文件中每個數據求冪(子菜單第9項);格點文件中每個數據對另一個格點文件對應項進行平方和、平方差、求平均操作(子菜單10/11/12項)
格點文件中每個數據求絕對值(子菜單第13項)
操作完畢后會輸出新的格點文件,新的格點文件可繼續使用gsgrid進行操作。見例4、例5。
第六類功能(程序中第11-12項):第11項功能為提取某個等值面上的格點。第12項功能能夠將代表某個屬性的格點文件A投影到格點文件B的數值所定義的等值面上,輸出文件中包含這些等值面上的格點的坐標和A格點文件中坐標相對應的格點的數值。見例6、例7。
3. 例子
以下例子建議初學者從頭看一遍。
例1:得到空間中每個坐標點的靜電勢
用高斯計算某個體系,寫明%chk,然后用formchk將chk轉換為fch,例如得到marioholic.fch。cubegen.exe在高斯目錄下,將cubegen程序與marioholic.fch放到同一個文件夾,在dos中進入相應目錄下運行:
cubegen 0 potential marioholic.fch aneta_langerova.cub 0 h
就得到了靜電勢格點文件aneta_langerova.cub。
(也可以在這個文件夾下用文本編輯器建立一個批處理文件,比如a.bat,內容就是上面這條命令,然后雙擊運行a.bat)
運行gsgrid,比如輸入:(注:下文//后面皆代表注釋,不要輸入。換行處就是敲回車)
c:\aneta_langerova.cub //若.cub文件與gsgrid在同目錄,只需輸入文件名)
1 //功能1
即在相同目錄下得到output.txt。里面包含aneta_langerova.cub里每個數據點的數值及其坐標。
------------------------------------
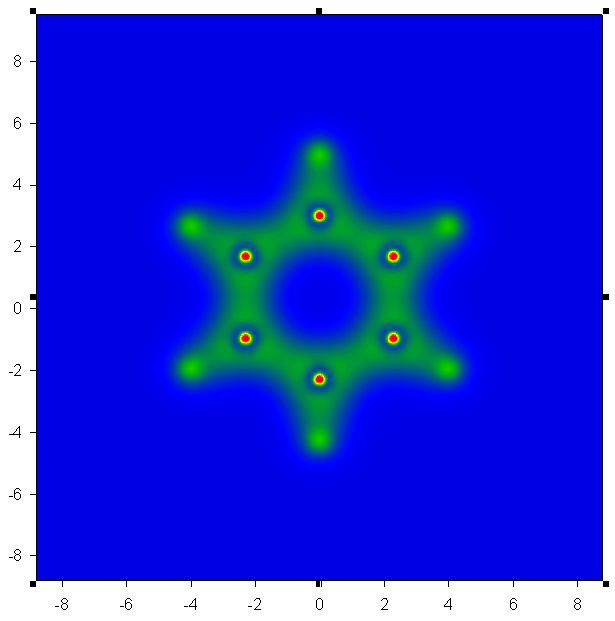
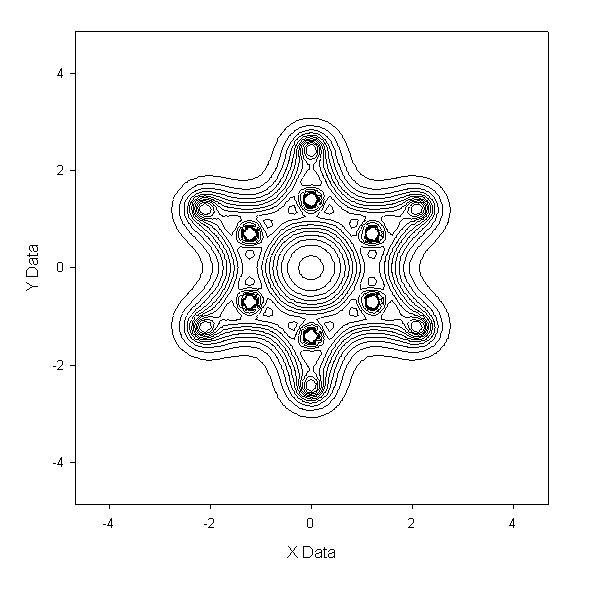
例2:畫Z=0平面的電子密度圖
先得到電子密度格點文件,運行:cubegen 0 density mizuki_nana.fch otoboku.cub -4 h
運行gsgrid,輸入:
c:\otoboku.cub
2 //功能2,提取某XY平面的數據
0 //設定此XY平面的Z值為0
得
到output.txt,和例1一樣。由于格點文件數據是離散的,故這里面的數據點只包含.cub中的Z值最接近于0的那個平面中的數據點。然后可以放到
sigmaplot等軟件做圖。以苯分子為例子(壓縮包內c6h6.fch),以此法作圖得到benzene-density.PNG及benzene-
density-contour.GIF
再比如想得到X在-3.3至5.2范圍內的YZ平面上的數據點的平均值,可使用程序第6個功能,運行gsgrid,輸入
c:\otoboku.cub
6 //功能6
-3.3 5.2 //Z值范圍
------------------------------------
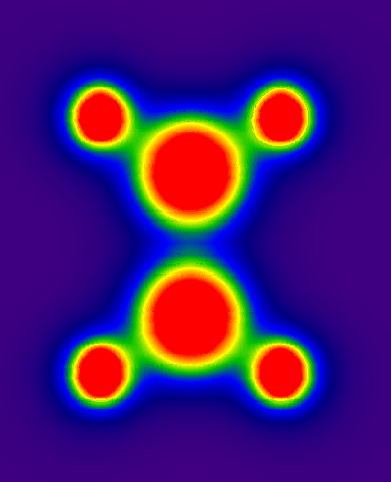
例3:得到三個原子定義的平面的靜電勢圖
設某個分子有三個原子編號為3、6、7,想得到這三個原子定義的平面上的靜電勢圖,但是這三個原子并不平行于XY/YZ/XZ,故不能用程序的2、3、4項功能通過例2的方法做圖。在V1.3版中添加了任意平面做圖可以實現這個目的。
cubegen 0 potential ethane.fch 1.cub -4 h //ethane.fch在附件中,這里使用fine格點,否則圖像會破碎,見后面說明。
運行gsgrid,輸入:
c:\1.cub
8 //功能8
3,6,7 //輸入三個原子序號
0.014 //以0.014埃為格點-平面臨界距離
y //將平面數據通過旋轉投影到XY平面上
這樣得到的結果就像例2,數據點z值皆為0,可以直接用繪圖軟件做圖,使用sigmaplot得到的結果如附件中ethane-PES.PNG所示。
再比如想自定義三個點得到定義的平面的數據,運行gsgrid,輸入:
c:\1.cub
9
2.3,1.1,2.55 //第1個點的XYZ
0.23,1.33,4.5 //第2個點的XYZ
0,-1.29,3.11 //第3個點的XYZ
0 //用默認的格點-平面臨界距離
n //不投影至XY平面
------------------------------------
例4:得到某體系第4個分子軌道在某XY平面的密度分布
首先需要得到分子軌道波函數格點文件,運行cubegen 0 mo=4 divokej_bill.fch 1.cub 0 h。
求每個格點數據的2次方,因為波函數平方為概率密度。運行gsgrid,輸入:
c:\1.cub
10 //第10項功能,即計算功能
9 //子菜單第8項功能,求冪
2 //設定指數,輸入的數除了整數外,也可以是小數、負數。要注意底數是負數時指數須為整數,否則結果為NaN。
z.cub //輸出的格點文件,不寫路徑則生成在當前路徑下
假設這個分子軌道由兩個電子占據,故每個格點數據應再乘以2,再次啟動gsgrid,輸入:
z.cub
10 //第10項功能,即計算功能
5 //子菜單第5項功能,乘以一個常數
2 //每個格點數據乘以2
lusaint.cub //輸出的格點文件
最后再啟動gsgrid,提取lusaint.cub的某XY平面數據即可,見例1。
再例如我們想得到mo=4和mo=5兩個分子軌道上占據的電子密度和的格點文件。mo=4的格點文件仍是上面的1.cub,mo=5的格點文件是2.cub。運行gsgrid
1.cub //先輸入第一個格點文件
10 //第10項功能,即計算功能
10 //子菜單第10項功能,求平方和,即A^2+B^2
2.cub //第二個格點文件
Bakuretsu_Tenishi.cub //輸出的格點文件
若每個軌道是雙占據的,再把Bakuretsu_Tenishi.cub用gsgrid乘以常數2即可。
實際上用gsgrid把1.cub和2.cub單獨求平方,然后再相加得到的結果和上面一樣,這里用子菜單第10項功能是因為比較方便,只需要一步而不必三步。
要注意,兩個格點文件間運算,兩個格點文件必須是相同體系,有相同的格點數目。
------------------------------------
例5:多分子軌道格點文件的讀取
每
個高斯格點文件可以包含多個軌道,從gsgrid
V1.5開始支持這樣的格點文件。這里仍然使用例4中的第二個例子,需要獲得MO=4和MO=5的總電子密度格點文件。為了方便,我們現在把mo=4和
mo=5的格點都寫進一個文件里,例如cubegen 0 mo=4,5 bitboys.fch sob.cub 0 h。運行gsgrid,輸入
sob.cub
1 //選取的格點文件若包含多個軌道信息,則會出現選擇軌道的提示。這里輸入1代表選擇第一條軌道,即MO=4
10 //第10項功能,即計算功能
10 //平方和運算
sob.cub //第二個格點文件,因為MO=5的軌道就在sob.cub中,所以仍用sob.cub
2 //選格點文件中第二條軌道,即MO=5
z.cub //輸出文件
閉殼層體系時再把z.cub的內容乘2即可。這樣將多條軌道合并在一個格點文件中用起來比較方便,但是會造成格點文件成倍增大,gsgrid讀取也會變慢。
第一個格點文件和第二個格點文件可以包含的軌道數目不同,也可以不是一類內容,如分子軌道與電子密度值,只要格點在數目、空間位置上是對應的即可。
使用gsgrid也可以提取多分子軌道格點文件中某一個軌道的格點文件,即選擇那條軌道,使用比如加0或者乘以1的運算。
高
斯的分子軌道(包括多分子軌道)的格點文件與其它類型格點文件的主要區別在于前者的原子數為負,在原子坐標后面會加入分子軌道信息(見后面附帶的格點文件
格式說明)。如果第一個格點文件是分子軌道格點文件,則運算后生成的新格點文件也將是分子軌道類型。若格點文件中原子數為正/負,則必須無/有分子軌道信
息,否則格點文件無法被gsgrid正確處理。
------------------------------------
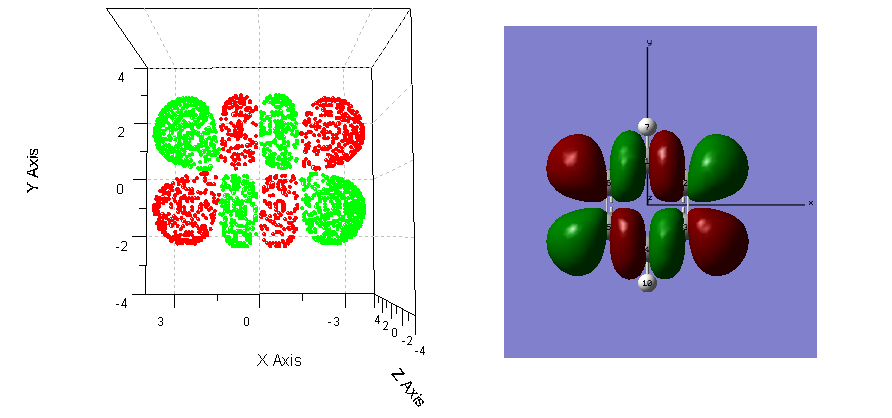
例6:提取某個等值面的數據點
這個例子要用到1.6加入的功能11。下面我們要得到自帶的test文件夾下c6h6.fch體系(苯,3-21G)的第19個分子軌道波函數的絕對值為0.02的等值面的格點。首先得到第19個分子軌道的格點文件:
cubegen 0 mo=19 c6h6.fch c6h6-mo-19.cub -3 h
使用功能11、12時最好使用中等及以上精細度的格點文件,所以這里使用-3參數即中等精細度。
運行gsgrid,依次輸入:
c6h6-mo-19.cub
11 //第11項功能,提取某個等值面的格點
0.02,0.02
//這兩個數分別定義下界和上屆的數值,如果某格點的數值在這個范圍內,則這個格點就會輸出到output.txt。這里上界和下屆輸入的數值一樣,則程
序會將這個數值的±3%作為上下界,即提取0.0194至0.0206范圍內的格點(若不允許有±3%的偏差,得到的數據點太少)。
現在得到了output.txt,但這里只有分子軌道波函數為正值的部分,我們還想得到等值面為-0.02的格點,故把剛才得到的output.txt備份,再次在gsgrid中運行上述命令,0.02,0.02替換為-0.02,-0.02即可。
現在我們將這些格點放到origin里面做三維散點圖,正值與負值部分分別用紅色和綠色表示,得到了isosurface.PNG文件的左圖。可以看到,我們提取的等值面上的格點是正確的,形狀與gaussview中顯示的此分子軌道波函數的0.02的等值面完全一致。
------------------------------------
例7:將靜電勢投影到電子密度為0.001的等值面
首先分別得到電子密度和靜電勢的格點文件,注意它們必須擁有相同格點數,每個格點坐標相等。
cubegen 0 density c6h6.fch c6h6-m-density.cub -3 h
cubegen 0 density c6h6.fch c6h6-m-eps.cub -3 h
運行gsgrid,依次輸入:
c6h6-m-density.cub //通過這個格點文件確定等值面的格點
12 //選功能12
0.001 //等值面數值為0.001
4 //設定上下限偏差,用百分數表示。這里即設定數值在0.001±4%范圍內的格點看做等值面格點
c6h6-m-eps.cub //將這個靜電勢格點文件投影到剛才設的等值面上并輸出
這樣我們得到的output.txt中的數據點的坐標就是c6h6-m-density.cub文件中數值為0.001±4%范圍所定義的等值面上的格點的坐標,而這些點的數值就是c6h6-m-eps.cub文件中坐標相對應的點的數值。
------------------------------------
關于功能8和功能9的細節說明:
由
于格點文件的數據點在空間上是規律排布的,若自行定義一個平面,數據點顯然不會恰好在平面上。所以想得到某個平面上的數據,就必須將平面附近的數據點投影
到平面上。在gsgrid中,只有數據點與平面垂直距離小于“格點-平面臨界距離”的數據點才會被投影,符合要求的數據點投影的方法是沿著法線方向移動至
平面。格點-平面臨界距離的設定會影響實際效果,一般用默認值即可(輸入0使用建議值)。
有時通過這種投影方法獲得的某平面的數據在做等值 線圖時會發現圖像雖然形狀正常,但略有破碎,或者個別點數據怪異。此時可降低格點-平面臨界距離來解決,例如改為默認值的1/2、1/4再重新做圖,但是 格點-平面臨界距離如果太近的話,投影到平面上的格點數太少,圖像會變得粗糙。最好的解決方法是生成格點文件時使用fine格點,此時格點密度加大,投影 到平面上的數據準確度較好,可消除圖像破碎問題,此時建議將格點-平面臨界距離設為推薦值的1/2。
有時得到的等值線圖有棱有角,是因為格點文件不夠精細造成,亦可通過使用fine格點解決,也可以通過改變等值線或色彩范圍、調節格點-平面臨界距離來得到一定改善。
將平面的數據轉移至XY平面的過程,是以自定義平面與XY平面的交線為軸,旋轉自定義平面來實現的。不會使平面上數據得到的圖像有任何變形,相當于眼睛垂直于自定義平面觀看。
如果定義的平面與XY/YZ/XZ平面平行,程序會予以提示,此時請分別用2、3、4項功能代替,以得到更好結果。
關于8、9項功能使用若有問題或結果異常,請E-mail來信。
4. 使用注意
使用cubegen時,末尾一定要用h參數。
cubegen倒數第二個參數可以是-2,-3 和-4 分別對應于關鍵字Coarse,Medium和Fine。用了fine文件會很大,gsgrid讀取也比較慢,但相應地數據點更精細,建議繪制文獻圖時使用fine。若設為0用默認值,大約與Medium相當。
gsgrid目前只支持X、Y、Z平移向量與坐標軸X、Y、Z平行的格點文件,即一般情況下的格點文件。
格點文件中所用單位為波爾半徑,等于0.529177249埃,讀入VMD、gaussview后會被轉換為埃。為了與可視化程序保持一致,本程序在讀入格點文件時已經將單位轉換為埃,輸入參數和輸出結果都以埃為單位。
5. 使用技巧:
若想使程序以silent模式運行,也就是敲一個命令即完成指定任務,不必每一步都手動輸入,可以編輯一個文件,比如batch.txt,內容如下:
1.cub
8
3,6,7
0.014
y
[空行]
[空行]
這樣只要運行gsgrid < batch.txt,就可以完成例3中的任務,每一步都根據batch.txt的內容自動輸入了。如果處理很多格點文件,這樣會方便很多。
6. 附錄
Gaussian格點文件格式簡介:
例如水的靜電勢的格點文件
Title Card Required potenial //Title,不被gsgrid處理
Electrostatic potential from Total SCF Density //Title,不被gsgrid處理
3 -4.970736 -4.970736 -4.761332 //原子數(如果是分子軌道格點文件原子數為負值) 原點的X/Y/Z坐標
80 0.125841 0.000000 0.000000 //第一個“坐標軸”上有80個數據點,每個數據點間隔為0.12584波爾半徑
80 0.000000 0.125841 0.000000 //第二個“坐標軸”
80 0.000000 0.000000 0.125841 //第三個“坐標軸”
8 8.000000 0.000000 0.000000 0.209404 //原子序號(氧),電子數,X/Y/Z坐標
1 1.000000 0.000000 1.481500 -0.837616
1 1.000000 0.000000 -1.481500 -0.837616
//若是分子軌道格點文件,這里有分子軌道信息。第一個數字為格點文件包含的分子軌道的數目,接下來是每個分子軌道的序號。例如3 1 5 7就是代表格點文件含3個分子軌道,分別是1、5、7號。分子軌道信息內容中每10個數字換一行。
7.33384E-03
7.33602E-03 7.33151E-03 7.31988E-03 7.30070E-03 7.27354E-03
//每個點的數據,每六個換一行,E13.5格式。如果數據點數不是6的倍數,最低維循環后不足6個數據位置的地方會留空。
7.23796E-03 7.19353E-03 7.13981E-03 7.07636E-03 7.00279E-03 6.91867E-03
6.82364E-03 6.71732E-03 6.59939E-03 6.46955E-03 6.32753E-03 6.17312E-03
......
這些數據點數據輸出的循環次序是:最低維=第三個“坐標軸”(此例即是Z軸),中維=第二個“坐標軸”,最高維=第一個“坐標軸”。
數據點與其對應的坐標關系是:
a(i,j,k)%x=originx+trans1x*(i-1)+trans2x*(j-1)+trans3x*(k-1)
a(i,j,k)%y=originy+trans1y*(i-1)+trans2y*(j-1)+trans3y*(k-1)
a(i,j,k)%z=originz+trans1z*(i-1)+trans2z*(j-1)+trans3z*(k-1)
trans1/x/y/z代表第一個“坐標軸”三個分量,(或曰:格點的平移矢量)
trans2/x/y/z代表第二個“坐標軸”三個分量
trans3/x/y/z代表第三個“坐標軸”三個分量
originx/y/z代表原點位置
i、j、k代表某點在各個“坐標軸”上的數據點序號。
如果用cubegen默認設置,一、二、三號坐標軸實際上就相當于笛卡爾坐標軸X/Y/Z,彼此正交。
如果手動設置網格,可以自定義“坐標軸”的方向,不平行于笛卡爾坐標軸,但這種情況gsgrid只能提取全部數據點(第一個功能),不能提取指定平面的數據點。
也可以用cube、prop關鍵字生成格點文件,參見高斯手冊,格式與cubegen生成的一樣,但更建議使用cubegen來做。