使用GIMIC計算和分析磁感應電流密度
2019-Jun-22注:由于后來出了好用得多的GIMIC 2.0版,此文介紹的GIMIC老版本的程序使用部分的內容已經完全過時了。想用GIMIC研究問題的讀者一定要仔細看《考察分子磁感生電流的程序GIMIC 2.0的使用》(http://www.shanxitv.org/491),其中還包含24分鐘演示視頻。
使用GIMIC計算和分析磁感應電流密度
文/Sobereva @北京科音 2012-Jul-15
1 GIMIC簡介
分子中磁感應電流密度的基本概念以及使用AICD程序繪制磁感應電流密度的方法在《使用AICD程序研究電子離域性和磁感應電流密度》(http://www.shanxitv.org/147)中已經介紹過。然而,使用AICD程序有一些缺點:
1 必須有Gaussian的源代碼版權而且會編譯Gaussian源碼
2 AICD是用C++寫的,而且寫得很復雜,注釋和很多變量名都是德文,難以自行修改
3 AICD不能直接輸出每個位置的感應電流密度數值,因此沒法利用這些數據按自己所愿去寫程序進一步分析,比如作平面圖、對某個截面進行積分
4 AICD利用的是Gaussian的CSGT方法的磁性質計算的輸出信息,而CSGT的結果隨基組收斂速度比常用的GIAO方法要慢。
使用GIMIC程序分析磁感應電流密度則可以避免上述問題。
GIMIC程序實現的是GIMIC(gauge including magnetically induced current method)方法。GIMIC方法基于GIAO(gauge including/invariant atomic orbitals,范規不變的原子軌道)方法,并依靠磁性質計算過程產生的密度矩陣和密度矩陣對磁場的導數矩陣來計算感應電流密度,計算公式見下文提到的原文、綜述和幻燈片,這里就不介紹了。雖然在GIMIC程序里也要做不少運算,但還是很快的。相對來說,AICD程序中計算電流密度的過程就簡單多了,就是直接將CSGT計算中間生成的積分點上的電流密度張量投影到均勻分布的笛卡爾坐標點上,然后乘上外磁場向量就得到了電流密度。
原則上,GIMIC程序可以支持任何使用GIAO方法計算磁性質的量子化學程序。不過,目前版本GIMIC程序只支持CFOUR、ACESII和Turbomole程序。Turbomole大概賣2W RMB,如果不想花錢的話,就用免費開源的CFOUR,借助它可以在HF以及高精度耦合簇級別下計算電流密度,但遺憾的是不支持DFT。關于CFOUR的基本用法我在《CFOUR程序的編譯和使用方法簡介》(http://www.shanxitv.org/150)一文中已經介紹了,本文也只討論使用CFOUR時的情況。ACESII和CFOUR是同根生,功能很大程度相似,而且還支持DFT,但這程序非常難對付,沒頑強毅力的話還是別指望能馴服它。
GIMIC可以輸出一個空間范圍或指定平面上均勻分布的格點上的電子密度、電流密度及其模和散度,也可以輸出自定義的一批點上的這些性質。還可以對電流密度在指定的平面上做積分,得到比如穿過相鄰兩個原子的凈電流。比起AICD來說這個程序更為靈活,專業。GIMIC絕大部分使用Fortran90編寫,代碼改起來也相對比較容易。
GIMIC的缺點是手冊寫得太簡單且含糊不清,很多地方必須靠猜或者反復嘗試才能搞懂,有些關鍵詞都沒有介紹,代碼有的地方不完整有bug。雖然GIMIC方法從形式上能拆成各個軌道的貢獻,但是目前版本的程序還不能像AICD程序那樣分解成軌道的貢獻。
GIMIC是免費開源的,可以從此處下載https://gitorious.org/gimic。首先要注冊個用戶名,然后在這個頁面中點source tree按鈕,然后在右側點Download master as tar.gz來將目錄中的文件打包下載。(注:網址已經失效,可在此下載/usr/uploads/file/20160121/20160121171602_29686.rar)
GIMIC方法的原文見JCP,121,3952,一個很不錯的介紹GIMIC及其應用的綜述見PCCP,13,20500。在寡人制作的幻燈片《Utilizing AICD and GIMIC programs to study magnetically induced current density》當中也簡要介紹了感應電流密度、磁性質的計算、CFOUR、AICD及GIMIC程序的基本原理和用法,可以到這里下載http://www.shanxitv.org/148(個別地方的介紹與本文不同,以本文為準)。
除了前面已提及的AICD和GIMIC,還有一種CTOCD-DZ(continuous transformation of origin of current density-diamagnetic zero)方法也被不少研究者用于繪制感應電流密度。然而只有修改版的SYSMO程序能實現此方法,而此程序又不公開,所以這里就不多提它了。
PS:可能有讀者對GIAO、CSGT這些概念不清楚,關于磁性質計算的方法這里簡要提一下。在計算磁性質時,磁場B并不是直接出現在薛定諤方程中的,薛定諤方程中引入的是B對應的矢量勢A,即原先的動量算符p被替換為π=p-qA,q是指粒子的電荷(對于電子q=-1)。A與B的關系為B=▽×A。由于任何標量勢C的梯度的旋度都是零,因此A有一定任意性,將A替換為A'=A+▽C在原理上不會影響體系性質的計算結果,這被稱為“規范變換”。等價地可以表述為A(r)=(1/2)B×(r-R_o),這里r是某點位置,R_o稱為“規范原點”,它是人為任意選取的。當基組是完備的情況下,R_o的選取不會影響磁性質計算結果,但在實際的有限基組計算下,它的選取就會影響結果,所以得想辦法消除磁性質計算中這個任意性問題,以滿足“規范不變性”,使磁性質(包括電流密度)能唯一地獲得。怎么解決測量原點任意性問題,導致了不同磁性質計算方法,GIAO是絕大多數量化程序(包括Gaussian)默認的計算磁性質的方法,它在一般量化計算用的高斯函數中引入了復數因子,使計算出的各種性質不依賴于測量原點的選擇(而此時波函數自身則依賴于原點)。另外的一些辦法是對每個定域化軌道用單獨的測量原點以盡可能減小誤差,比如IGLO(individual gauge for localized orbitals)和LORG(localized orbital-local origin)方法。IGAIM(Individual Gauge for Atoms In Molecules)和之后的CSGT(Continuous Set of Gauge Transformations)的本質十分類似,計算中使用了多重測量原點。
GIAO和CSGT在小基組時,比如6-31G*級別下結果差得不少,絕對屏蔽值能差出幾十ppm。而當基組特別大時,兩種方法結果就非常一致了。因此計算NMR等磁性質時,應當注明是用的什么方法算的。CSGT的結果隨基組收斂比GIAO慢,一般還是用GIAO為好,可參見JCP,104,5497的討論。
2 修改GIMIC代碼
下載之后先解壓壓縮包。里面有fortran源代碼文件和很多腳本文件。程序的手冊在壓縮包里Documentation目錄下。
在GIMIC程序輸出的cube文件格式不對,沒法被主流的可視化軟件所讀取,這是因為GIMIC生成的cube文件不滿足標準cube格式的換行要求,而且用的浮點格式也不是標準的1PE13.5,而且cube文件里沒有分子坐標信息。因此,需要對代碼進行一些修改,也使得生成的cube文件里總有原子坐標信息,其內容是我們隨便設的,是一個位于原點的氫原子。
在cubeplot.f90里搜索
do i=1,npts(1)
do j=1,npts(2)
do k=1,npts(3)
write(fd,'(f12.6)',advance='no') pdata(i,j,k)
if (mod(l,6) == 5) write(fd,*)
l=l+1
end do
end do
end do
將之改為
do i=1,npts(1)
do j=1,npts(2)
write(fd,"(6(1PE13.5))",advance="no") pdata(i,j,1:npts(3))
write(fd,*)
end do
end do
把cubeplot.f90的35行的0改為1
把cubeplot.f90的38行后面加上write(fd,"(i5,4f12.6)") 1,1D0,0D0,0D0,0D0
把jfield.f的610行后面加上
if (mod(l,6) /= 5) then
l=0
write(fd1,*)
write(fd2,*)
end if
在jfield.f里搜write(fd, '(i5,3f12.6)') 0, origin,將0改為1
在jfield.f里搜write(fd, '(i5,3f12.6)') npts(3), 0.d0, 0.d0, step(3),在后面加上write(fd,"(i5,4f12.6)") 1,1D0,0D0,0D0,0D0
在gimic.in里面把print "Error: either spacing or grid_points must be specified"這一行后面的sys.exit(1)這行刪掉,否則無法令程序計算自定義的一批點上的電流密度等屬性。
GIMIC源程序(2012年6月5日下載的)和按上述過程改好的文件可以從這里下載:/usr/uploads/file/20150610/20150610050217_74417.rar。
3 編譯GIMIC代碼
首先確保CFOUR已經編譯好,并且編譯時帶著--enable-gimic選項,這將會編譯出xcpdens程序用于輸出后文提到的XDENS文件,這些都已在《CFOUR程序的編譯和使用方法簡介》中提到。
./configure FC=ifort (不寫FC=ifort則默認用gfortran,也能正常編譯。如果要指定安裝目錄而非默認的/usr/local/bin,加上比如--prefix=/sob/gimic)
把Makefile的INST_PROGS+=xcpdens這行注釋掉
make
這將在GIMIC目錄下生成gimic.x,這是GIMIC程序的計算模塊。
將GIMIC/tools/MOL2mol.sh打開,將ACES2變量改為實際路徑,如ACES2=/sob/cfour_v1,將后面兩處$ACES改成$ACES2。
將GIMIC/tools/xgimic2.sh打開,將ACES2變量改為實際路徑,如ACES2=/sob/cfour_v1
運行make install。這將把gimic.x和一些腳本拷貝到/usr/local/bin里
自行將GIMIC/tools/xgimic2.sh拷到/usr/local/bin里
(注:按照GIMIC官方的說法是先按普通方式編譯CFOUR/ACESII,然后在GIMIC的./configure后面加上諸如--with-aces2=/sob/cfour_v1/lib,這樣就會編譯出xcpdens,但實際上GIMIC的configure腳本有問題,認不出編譯好的CFOUR/ACESII的靜態庫來。在編譯CFOUR時加上--enable-gimic也會編譯出xcpdens,是完全等效的。所以建議還是在CFOUR編譯時就用--enable-gimic選項編譯出xcpdens)
4 運行GIMIC
運行GIMIC需要三個文件:
1 mol文件,包含分子坐標和基組信息。
2 XDENS文件,包含密度矩陣和密度矩陣對磁場的導數。
3 gimic.inp文件,里面的關鍵詞設定要讓GIMIC干什么。
mol和XDENS都由CFOUR的NMR計算來輸出。這里以使用GIMIC分析苯的感應電流密度為例介紹一下流程。首先建立一個空文件夾,編寫一個CFOUR的NMR任務的輸入文件,命名為ZMAT放入其中,內容如下
BENZENE
X
C 1 RCC
C 1 RCC 2 A60
C 1 RCC 3 A60 2 D180
C 1 RCC 4 A60 3 D180
C 1 RCC 5 A60 4 D180
C 1 RCC 6 A60 5 D180
H 1 RXH 2 A60 7 D180
H 1 RXH 3 A60 2 D180
H 1 RXH 4 A60 3 D180
H 1 RXH 5 A60 4 D180
H 1 RXH 6 A60 5 D180
H 1 RXH 7 A60 6 D180
RCC=1.39864
RXH=2.48607
A60=60.0
D180=180.0
*CFOUR(CALC=HF,BASIS=6-31G*,PROPS=NMR,SYMMETRY=ON)
[空行]
注意必須寫上SYMMETRY=ON,盡管這是默認的。
運行xgimic2.sh --scf |tee cfour.out
如果用的是微擾方法,--scf應該改為--mbpt,如果是耦合簇,用--cc。xgimic2.sh會依次調用CFOUR的各個模塊進行計算,最后當前目錄下就有了XDENS文件。這個文件是文本文件,假設有66個基函數,那么里面就有66+66*3個數,貌似前66個是密度矩陣,后3*66個是它對磁場三個方向的導數矩陣,每個矩陣末尾空一行,因此總共多出4行。
(注:由于xgimic2.sh調用CFOUR計算時是單進程模式,故沒法照常以MPI方式并行運行CFOUR。但如果編譯CFOUR時用了MKL,則也能依靠MKL來并行,也就是先執行export MKL_NUM_THREADS=n,n是當前能用的所有核心數)
接下來直接輸入MOL2mol.sh
這步將會新建個臨時目錄并把ZMAT拷進去,將SYMMETRY從ON變為OFF,并重新調用CFOUR迅速過一遍,生成無對稱性的mol文件并復制到當前目錄下,其中包含了所有原子的坐標和基組。
(注:帶著對稱性計算時xgimic2.sh那步已經產生了MOL文件,但其中只有對稱唯一部分的坐標。如果輸入文件寫的是SYMMETRY=OFF了,則可以省略這步,直接將MOL改名為mol即可)
把gimic.inp從GIMIC/examples里面拷過來,在此基礎上進行修改。
此時gimic.inp、mol、XDENS三個文件都有了,直接輸入gimic即開始計算。(gimic是個腳本,已經被拷到了/usr/local/bin)
這里順便說一下,對于閉殼層體系的電流密度計算,用HF/6-31G*就能得到不錯結果。對于開殼層體系,電子相關必須著重考慮,此時應當用更高級別方法結合更大一些基組。基組建議用3-zeta加極化的,理論方法用CCSD最穩妥。微擾方法不要用,往往存在厲害的自旋污染問題。DFT是否適用還沒有經過大量檢驗,如果用的是turbomole或ACESII倒是可以試試。
5 GIMIC的輸入文件
GIMIC輸入文件gimic.inp中的每個關鍵詞的含義在手冊中都有說明,這里有重點地說明一下。首先注意輸入文件中所有長度單位都是Bohr。
輸入文件中第一部分是全局設定,典型的是如下這樣,#后面是自帶的注釋
dryrun=off # don't actually calculate (good for tuning grids, etc.)
mpirun=off # run in parallel mode
title=""
basis="mol" # Name of MOL file with coordinates and basis sets
density="XDENS" # File with AO density and perturbed densities
spherical=off # don't touch, unless you REALLY know what you are doing
debug=1 # debug print level
diamag=on # turn on/off diamagnetic contributions
paramag=on # turn on/off paramagnetic contributions
GIAO=on # turn on/off GIAOs. Don't change unless you know why.
openshell=false
screening=on # use screening to speed up
screen_thrs=1.d-8 # Screening threshold
show_up_axis=true # mark "up" axis in .xyz files
calc=[cdens, integral, divj]
這些參數中一般只需要自行設兩個,其余的都不用動。openshell選擇是否是開殼層計算,calc設定計算哪些任務。GIMIC包含這幾類任務:
cdens:計算指定區域內均勻分布的格點上的感應電流密度
divj: 計算指定區域內均勻分布的格點上的電流密度的散度
edens:計算指定區域內均勻分布的格點上的電子密度
integral:計算電流密度在某平面的積分。積分方式是二維高斯積分。
比如上面的例子,就代表依次做cdens、integral、divj這三個任務。
輸入文件的第二部分就要設定每類任務的具體選項,主要設磁場方向和格點分布。比如divj任務的選項就都在divj {...}里面夾著。
對于cdens、divj、edens任務,一般只需要設定以下參數。其它參數直接用模板輸入文件的默認設定就行了。
magnet_axis:設定磁場順著哪個軸,比如-y就是向著y軸負方向加磁場。
magnet:設定磁場矢量。比如[0.0, -1.0, 0.0]就和magnet_axis=-y的含義一樣。這個參數和magnet_axis只要設其一就行了,另一個應注釋掉。
grid(base) {...} 段落設定的是格點分布。其中
origin:設定格點原點
ivec和jvec:設定格點的前兩個方向的向量(i,j方向),第三個方向的向量(k方向)即是這兩個向量的叉乘。一般ivec和jvec分別設[1.0,0.0,0.0]和[0.0,1.0,0.0],此時i,j,k方向即是笛卡爾坐標軸的x,y,z方向。
lengths:設定格點空間范圍在三個方向上的總長度
spacing:設定在三個方向上的格點間距
grid_points:設定在三個方向上的格點數。它和spacing目的相同,故只能設其一,另一個要注釋掉。
對于integral任務,主要要設的是
magnet:設定磁場矢量
spin:考慮哪種自旋的貢獻
grid(bond) {...} 段落設定積分平面上積分點的分布方式,主要需要考慮的是
coord1和coord2:設定兩個點的坐標,積分平面就會在這兩個點之間,且垂直于其連線
bond:比如設[1,2],那么1號和2號原子的坐標就會分別作為coord1和coord2。當然了,bond和coord1/2只能同時設一個。
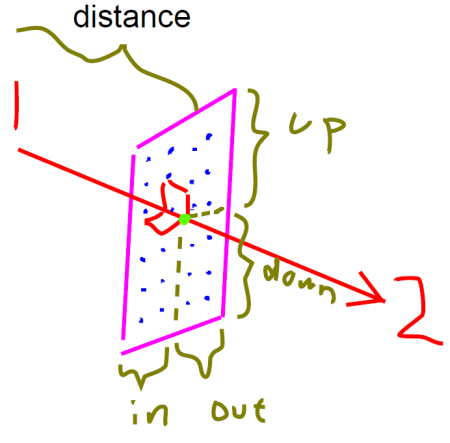
distance:積分平面距離第一個點的垂直距離
grid_points:比如設[30,30,0],那么在積分平面上就有30*30=900個積分點。最后一個值不用設,意義也不明。
spacing:設定積分點間隔。它和grid_points只能設一個,另一個要注釋掉
up、down、in、out:積分平面與coord1、coord2連線的交點根據此設定分別向四周擴展,就定義了積分平面的范圍。
用文字不容易說清楚積分平面的格點設定,我繪制了一張草圖。圖中紅色箭頭的兩端分別是coord1和coord2的位置。粉框是積分平面區域,垂直于紅箭頭,藍點是積分點,亮綠的點是積分平面與紅箭頭的交點。distance和up、down、in、out的含義標在圖上了。
6 GIMIC的輸出文件與可視化
首先注意,GIMIC輸出的文本文件里長度單位都是埃。本節所說的輸出文件的名字有的是在輸入文件中可調的,本文假設使用的都是模板輸入文件里的文件名設定。文件名前標星號的是重點,其它輸出文件可以無視。
本節的例子用的都是處在Z=0的XY平面上的苯分子,HF/6-31G*下計算,磁場順著Z軸正方向。i,j,k方向分別對應笛卡爾坐標x,y,z正方向。
6.1 cdens任務輸出的文件
grid.xyz:包含了分子坐標,并用8個X原子表示出格點數據范圍的8個頂點位置。(GIMIC輸出的各種xyz文件總是多出一個X原子,原因不明)
jtensor和jvector:是格點位置上的感應電流張量和感應電流向量,它們都是二進制文件。
*JVEC.txt和JMOD.txt:包含了由原點和i,j方向向量定義的平面上的電流密度矢量和電流密度模。
jmod.plt:是gopenmol格式的記錄了上述平面上的電流密度矢量的文件。prj_jmod.plt的意義不明。
*jmod.cube:是感應電流密度的模的格點數據,是diatropic和paratropic感應電流密度的模的總和,故總為正值。jmod_quasi.cube則是將diatropic和paratropic感應電流密度的模分別用正值和負值部分記錄。
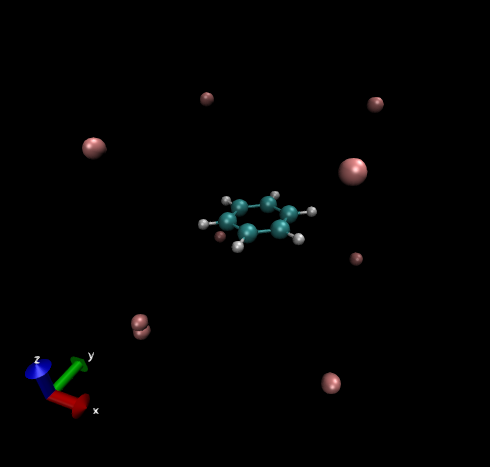
下面的圖是origin=[-8.0, -8.0, -8.0]、lengths=[16.0, 16.0, 16.0]、spacing=[0.2, 0.2, 0.2]格點設定時用VMD顯示grid.xyz時的圖像,肉色的X原子描述了格點數據的空間范圍,計算的點都落在以它們為頂點的長方體中,可見格點范圍涵蓋了整個分子。
電流密度是個矢量場,用一大堆箭頭描述三維空間各個位置的電流密度在分析時比較困難。而將每個點的電流密度矢量求模,就成為了張量場,可以通過觀看等值面直觀了解哪些位置電流密度比較大。
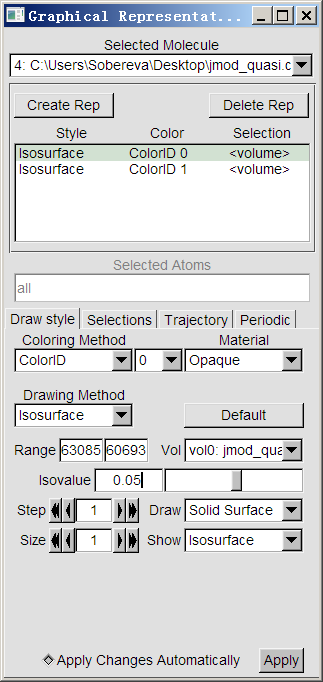
下面用VMD觀看電流密度的模的等值面,VMD可以在http://www.ks.uiuc.edu/Research/vmd/免費下載。啟動VMD,將上述格點設定下產生的jmod_quasi.cube拖進VMD main界面,選Graphics-Representations,將目前已有的Style通過點Delete Rep來刪掉。然后點Create Rep兩次創建兩個新的顯示方式,對這兩個顯示方式都按照下圖進行設定。兩個顯示方式的區別僅在于一個isovalue設為0.05且設為藍色以表現正值部分,另一個設為-0.05且設為紅色以表現負值部分
等值面顯示效果如下所示。藍色區域代表這部分有較大diatropic電流,也就是滿足左手定則的感生電流(即左手拇指朝向磁場方向時,左手其余手指彎曲時就指向感應電流方向,對純導體總是滿足)。這部分電流出現在苯環中部和外側,是由sigma軌道在苯環外側部分和pi軌道貢獻的。紅色代表這部分有較大paratropic電流,也就是與左手定則方向相反的感生電流,這是sigma軌道在苯環內側部分產生的。下面將更進一步分析。

接下來我們要利用JVEC.txt儲存的指定平面上的數據作苯的感應電流密度向量場圖。計算的平面區域與origin及lengths參數的關系如下圖所示,藍色是被計算的區域。當我們把k方向長度設為了0時,代表只計算二維平面的數據點,而不會輸出cube文件,這樣比較省時。
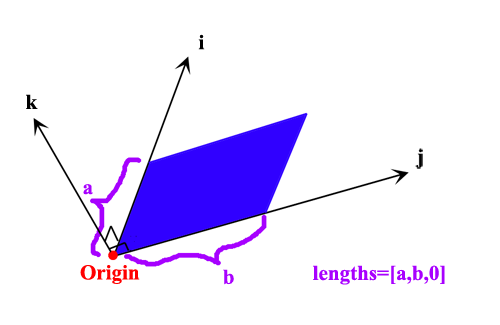
這里我們用這樣的格點設定:
ivec=[1.0, 0.0, 0.0]
ivec=[0.0, 1.0, 0.0]
origin=[-8.0, -8.0, 0.0]
lengths=[16.0, 16.0, 0.0]
spacing=[0.2, 0.2, 0.2]
這表明計算的平面范圍是Z=0的XY平面,即苯所在的平面,X和Y的范圍都是-8.0~8.0 Bohr,這兩個方向都是16/0.2+1=81個點。如果想計算分子平面上方1 Bohr的XY平面,就把origin設成[-8.0, -8.0, 1.0]。如果想計算的是個傾斜的面,顯然就得改ivec和jvec設定來調整i,j方向向量了。
JVEC.txt的內容是類似這樣的
...
2.2225444 0.5291772 0.0000000 -0.0198798 -0.0213785 0.0000000
2.3283799 0.5291772 0.0000000 -0.0156558 -0.0166937 0.0000000
2.4342153 0.5291772 0.0000000 -0.0117118 -0.0124604 0.0000000
2.5400508 0.5291772 0.0000000 -0.0083301 -0.0089750 0.0000000
2.6458862 0.5291772 0.0000000 -0.0056686 -0.0062921 0.0000000
...
前三列是每個點的xyz坐標,后三列是該點的電流密度矢量的xyz分量。通過sigmaplot、Origin、Surfer等支持繪制向量場圖的程序就可以將它繪制出來。這里通過Sigmaplot 11來繪制。
啟動Sigmaplot之后選File-Import-File,選JVEC.txt,點import。選Transform-Quick transform,運行col(7)=col(1)+2*col(4)和col(8)=col(2)+2*col(5)這兩個算式。這么做的目的是因為Sigmaplot作向量場圖需要每個向量的始端和末端的坐標,始端xy坐標就是JVEC.txt的前兩列,末端就是始端坐標加上電流密度向量。之所以算式中乘以2,是為了作出來的圖箭頭長度比較容易觀看,也可以事先將外加磁場矢量大小改為原來的2倍以達到相同目的。之后從左側的作圖類型列表里選vector plot(在最左列最下端),選XYXY模式,X1、Y1、X2、Y2分別選1、2、7、8列。作出圖后,放大到200%,然后雙擊圖,thickness設為0.1mm,angle in設為30,就能得到下面的圖像。
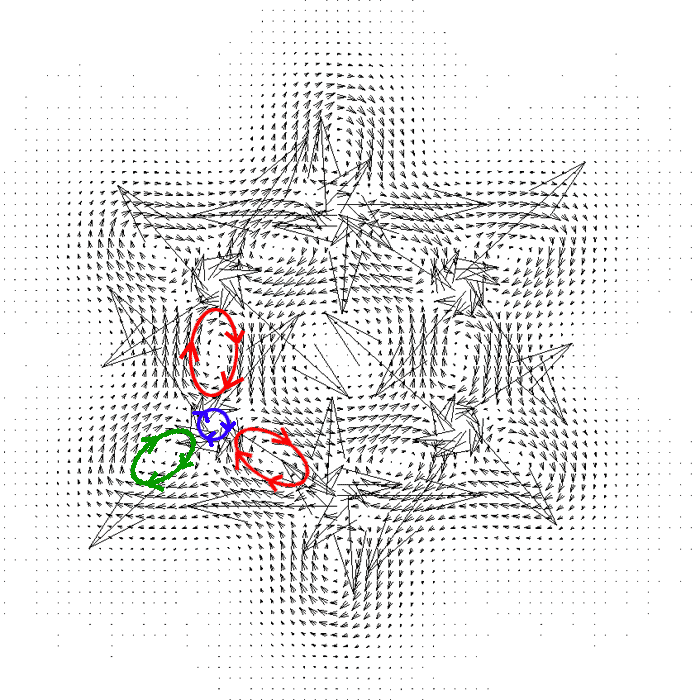
從箭頭方向上可以看出,每個C-C sigma鍵區域(紅圈所示)、每個C-H鍵區域(綠圈所示。由于氫原子沒有內核電子,所以整個氫與C-H鍵區域融為一體了)、每個碳原子核內核區域(紫圈所示,對應1s電子)都由于垂直朝向平面外的磁場形成了局部的環流,且在相應局部看都是diatropic電流。這些形成局部環流的區域都是電子定域性比較高的區域。
箭頭越長電流密度越大,可見碳原子內核電子的環流非常強,主要與離核很近區域電子密度很高且定域性很高有關。在原子核處,這環流會產生很強的朝平面內的磁場,抵消了很大一部分外加磁場,這就是核磁共振學中的磁屏蔽值的主要來由。
整體觀看這張圖,會看到苯環內側的感應電流是逆時針的,而苯環外側是順時針的,考察每個鍵的局部環流在環內側部分和環外側部分的方向就不難理解為什么會出現這種現象,這是各局部環流組合到一起所導致的。在苯環平面上,盡管內環有diatropic電流而外環有paratropic電流,由于它們的強度差不多,所以從分子外部來看這兩種方向的電流效應抵消了,因此光靠苯的sigma電子形成不了整體凈環流。對于其它多數飽和烷烴也是類似的,只靠sigma電子形成不了整體凈環流,表現出非芳香性特征。
將origin設成[-8.0, -8.0, 1.8896]后,用相同的步驟,就繪制出了苯環上方1埃(1.8896 Bohr)平面的電流密度圖。但注意由于這個平面上電流密度比較小,所以所做的變換應當是col(7)=col(1)+10*col(4)和col(8)=col(2)+10*col(5),這是令電流密度擴大更多倍數以使圖上箭頭長度合適。

由于此平面與sigma鍵軸有一定距離,所以這主要表現的是pi電子的效應。pi電子是在整個苯環上離域的,所以從圖中看到形成了整體的diatropic環流。由于sigma軌道產生的苯環的diatropic和paratropic環電流差不多抵消了,而pi軌道能產生較強的diatropic環電流,所以我們說的苯環的環電流是由diatropic所主導的,因此苯分子是芳香性分子。若是C4H4這種分子則是paratropic主導,就是反芳香性分子。如果整體看diatropic和paratropic電流大小差不多,因此無整體凈電流,就是非芳香性分子。
6.2 edens任務輸出的文件
edens:包含了分子坐標,并用幾個X原子表示出格點數據的各個頂點位置。
*edens.cub:格點空間內的電子密度數據的cube文件。
*edens.txt:包含了由原點和i,j方向向量定義的平面上的電子密度數據,是文本文件。通過它可以用sigmaplot等軟件繪制指定平面的電子密度圖。
EDENS:格點空間內的電子密度數據,是二進制文件。
6.3 divj任務輸出的文件
divj.xyz:包含了分子坐標,并用8個X原子表示出格點數據范圍的各個頂點位置。
divj.plt:是gopenmol格式的由原點和i,j方向向量定義的平面上的電流密度散度。
DIVJ:包括了空間格點范圍內電流密度矢量的散度,是二進制文件。
*DIVJPLT:是由原點和i,j方向向量定義的平面上的電流密度散度,是文本文件。前三列是笛卡爾坐標,第四列是散度值。
利用DIVJPLT文件通過sigmaplot等軟件可以將指定平面上的電流密度散度數據繪制出來。這里我們做苯分子平面上的圖。在前述格點設定下執行divj任務,將得到的DIVJPLT文件后綴名加上.txt,然后啟動sigmaplot,將此文件導入,選Transform-Quick transform,運行col(5)=abs(col(4)),也就是將散度數據取絕對值,這就不用考慮方向問題了,數值越大就對應于此處電流密度越發散,分析起來更方便。從左側的作圖類型列表里選Contour plot-Filled contour,選XYZ Triplet,X、Y、Z分別設為1、2、5列。作出圖后雙擊圖像,Scale里將start和end設為0和0.1,minor lines設為20,就得到了這樣的圖:
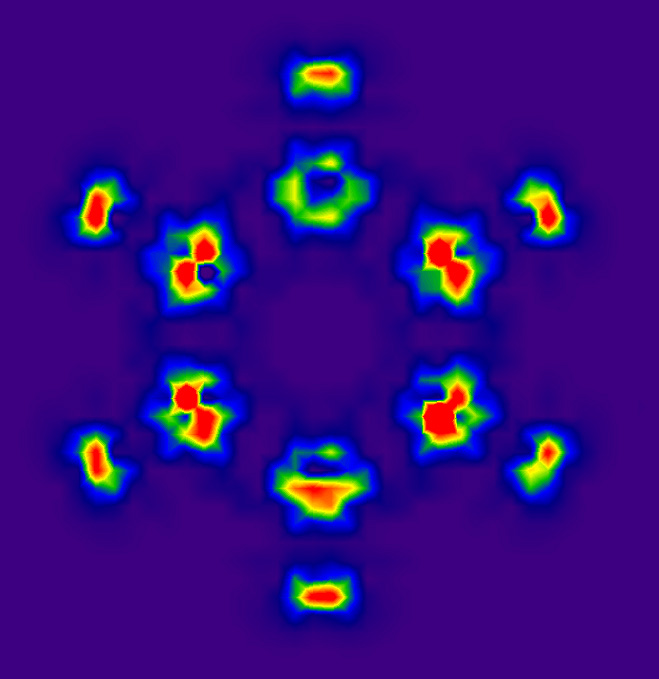
顏色越紅的位置電流密度發散性越強。對照前面的電流密度圖可看到,在sigma鍵局部環流中心處電流密度為0,四周電流圍著它打轉,電流沒有整體發散或收縮的趨勢,所以這些區域散度都為0。電流密度箭頭均勻并排前進的區域相當于均勻場,也理所當然地散度為0。在碳原子核附近的箭頭像旋轉灑水機噴出的水似的,很多地方散度非常大。由于散度圖對格點精度敏感(尤其是核附近),而當前的格點精度取得不高,而且格點分布不符合苯分子幾何對稱性,所以看起來圖像不細膩且不滿足對稱性,可以通過減小格點間距改善。
6.4 integral任務的輸出
這里用積分穿過苯的C1-C2鍵的電流密度為例說明。積分平面在C1-C2鍵的中點且垂直于鍵軸。格點設定如下
bond=[1,2]
distance=1.323 (半個C-C鍵鍵長)
up=6.0
down=6.0
in=2.0
out=6.0
這個任務會在屏幕上輸出諸如這樣的信息:
************************************************************
Induced current (au) : 0.461991
Positive contribution: 0.635794 ( 17.916310 )
Negative contribution: -0.173804 ( -4.897681 )
Induced current (nA/T) : 13.018629
(conversion factor) : 28.179409
************************************************************
這表明穿過這個平面的凈電流為0.461991 a.u.,乘上轉換因子28.179409(常數)折合于13.018629 nA/T。正值貢獻是0.635794 a.u.(17.916310 nA/T),通過前面的分析已經知道苯環的電流是diatropic主導的,也因此正值貢獻就是指的diatropic電流的積分。可見它的數值大小明顯超過paratropic電流產生的負貢獻。
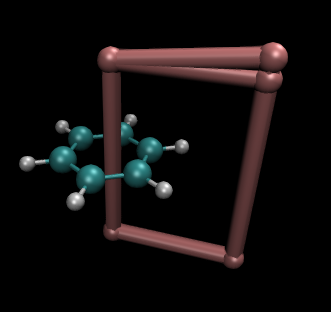
integral任務還同時輸出integral.xyz文件,它包含分子結構,并且通過4個X原子標出積分平面的區域。用VMD繪制它,令X原子相連,圍成的框就是積分區域,如下所示。當然,原則上積分點越密且積分區域越大積分精度越高。但是要注意別讓積分平面延伸到別的區域去。
7 自定義要算的數據點位置
如果想在一批自由設定的、沒有規律的位置上計算數據,就編輯一個文本文件,里面記錄要在哪些位置上計算(單位是bohr),可按照自由格式書寫,內容比如是下面這樣,文件名叫做extgrd
-3.5984053 -4.2334180 0.9999333
-3.4925698 -4.2334180 0.1199333
-3.3867344 -4.2334180 0.9999333
-3.2808989 -4.2334180 0.9999333
-3.1750635 -4.2334180 0.9999333
-3.0692280 -4.2334180 0.0
-2.9633926 -4.2334180 0.9999333
然后,將GIMIC輸入文件中相應任務類型內原先的格點設定部分注釋掉,而在其后新插入一行
grid(file) { file=extgrd }
之后照常使用GIMIC計算即可。例如對于cdens任務,輸出的JVEC.txt里就記錄了每個自定義位置的值。JTENSOR和JVECTOR記錄了這些位置的感應電流密度張量和感應電流向量,是二進制文件。