在VMD中計算RMSD衡量兩個結構間的幾何偏差
在VMD中計算RMSD衡量兩個結構間的差異以及疊合兩個結構
文/Sobereva@北京科音
First release: 2015-May-11 Last update: 2020-Feb-25
1 原理
RMSD(Root mean square displacement/deviation)搞分子模擬的人都很熟悉,但是搞量子化學的人可能好多都不知道。有人問怎么衡量激發態結構相對于基態的改變,雖然可以挨個比較鍵長鍵角的變化,但是若想用一個值衡量整體的結構改變程度,計算RMSD是最恰當也是最省事的,這里就簡單說一下怎么算。對任意兩個結構間都可以計算RMSD,比如可以衡量兩種極小點構型的差異、電子激發/電離/外加電場前后幾何結構的整體改變程度、形成復合物后單體結構的變化等等。
RMSD定義為
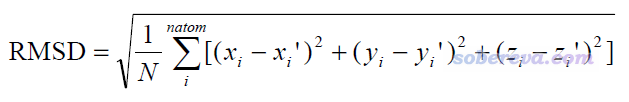
其中i循環所有原子,x_i和x_i'分別是第i個原子在第一個結構和第二個結構中的x坐標,y、z類似。
計算兩個結構間的RMSD之前必須先進行疊合(align),相當于令一個結構平移和旋轉來最小化兩個結構之間的RMSD,不做這一步的話計算出的RMSD是不能衡量兩個結構內部的差異的。
能計算RMSD、做疊合的程序不計其數,下面用免費的VMD程序(http://www.ks.uiuc.edu/Research/vmd/)的1.9.3版來演示,而且還將體現如何通過疊加圖直觀地展現兩個結構之間的差異。
2 實例:中性和陽離子狀態下的丙烷
本例演示如何計算丙烷在中性狀態下和在+1陽離子狀態下優化的結構之間的RMSD值,并且繪制出疊合后的圖。
首先把中性的丙烷用量子化學程序優化,把優化后的結構保存成VMD能認的一種格式,比如pdb、mol2、xyz等。這里我們用很常用的xyz格式,見《談談記錄化學體系結構的xyz文件》(http://www.shanxitv.org/477)。我們創建一個名為neutral.xyz的文本文件,把優化后的中性丙烷坐標拷進去,并在第一行寫上原子數,第二行是注釋行(內容隨便寫)。具體內容如下
11
Neutral
6 0.000000 1.277193 -0.259856
1 0.884678 1.321887 -0.907445
1 -0.884678 1.321887 -0.907445
1 0.000000 2.176504 0.367166
6 0.000000 0.000000 0.586502
1 0.877607 0.000000 1.247354
1 -0.877607 0.000000 1.247354
6 0.000000 -1.277193 -0.259856
1 0.000000 -2.176504 0.367166
1 -0.884678 -1.321887 -0.907445
1 0.884678 -1.321887 -0.907445
類似地也優化丙烷陽離子,把結構存為cation.xyz,內容如下
11
Ionized
6 1.249671 -0.343789 0.000081
1 1.315464 -0.947246 0.909976
1 1.314553 -0.947793 -0.909563
1 2.180924 0.290109 -0.000844
6 0.192643 0.673245 0.000031
1 0.022018 1.237089 0.919487
1 0.022792 1.237187 -0.919539
6 -1.442724 -0.286303 -0.000053
1 -2.107101 0.577835 -0.000705
1 -1.372956 -0.853783 -0.925484
1 -1.373233 -0.852316 0.926316
PS:如果你用的是Gaussian,把優化任務最后一幀的結構轉化為xyz格式最簡單的方法是用Multiwfn(http://www.shanxitv.org/multiwfn)。把Multiwfn的settings.ini文件里的iloadGaugeom設為1,然后用Multiwfn載入Gaussian優化任務的輸出文件,選擇主功能100的子功能2,就可以看見導出xyz文件的選項,選擇即可。
啟動VMD,把neutral.xyz和cation.xyz都拖到VMD Main窗口里載入。然后選擇Extensions - Analysis - RMSD Calculator,把左上角的文本框的內容改為all,取消選擇Backbond only復選框,然后點擊Align,在圖形窗口你會發現兩個結構已經疊合了,重疊度較高。然后再點RMSD按鈕,在RMSD Tool窗口下方顯示的Total RMSD就是兩個結構間的RMSD值。當前窗口看到的情況如下所示,可見RMSD是0.093埃。
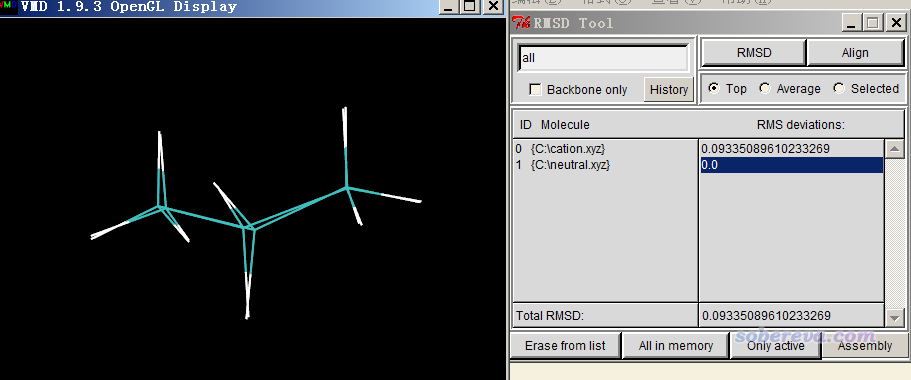
做疊合和計算RMSD的時候,我們可以只對指定部分進行。比如我們不輸入all,而是輸入比如serial 2 to 4 8,那么點Align按鈕時只會根據2、3、4、8號原子來做align,點擊RMSD按鈕時也只會計算這些原子間的RMSD。如果你對VMD的選擇語句不熟悉,建議參看《VMD里原子選擇語句的語法和例子》(http://www.shanxitv.org/504)。
VMD還可以把兩幀結構用不同顏色繪制出來便于直觀比較。做法是選Graphics-Representation,在Selected Molecule里先選擇對應中性的體系,把Coloring Method設為Color ID,在旁邊選擇13 Mauve,并把Thickness設為4使細線顯示得更粗。然后在Selected Molecule里選擇對應陰離子的體系,以類似操作把顏色設為藍色并加粗細線。最后在文本窗口輸入color Display Background white命令把背景改為白色,此時在圖形窗口會看到下圖,可見結構差異體現得十分清晰直觀

3 實例:不同的取代苯體系
有時候我們想對兩個主體結構相同,但某些部分不同的兩個分子的局部區域計算RMSD來衡量局部結構的差異。本例就用苯酚以及間硝基苯腈為例進行說明,二者在B3LYP/6-31G*優化后的結構如下
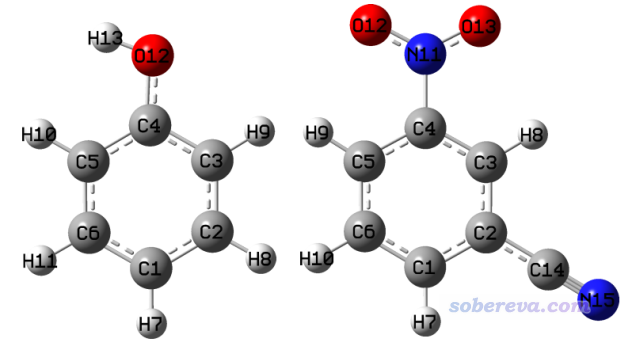
二者的差異在于苯環上連的基團。本例我們要讓兩個分子的苯環部分相互疊合(只需考慮碳原子)并計算RMSD。為了能夠正確疊合,一定要注意被疊合的部分要滿足兩個條件:(1)原子數相同 (2)原子順序一致。由上圖可見,這兩個體系中的苯環上的碳都是按連接順序排列的,所以可以直接做疊合和計算RMSD。如果其中一個體系的苯環上的碳是比如1,4,2,6,5,3這樣交錯排列的就沒法直接做疊合了,而需要在疊合之前自行編輯結構文件,調整原子順序使得條件(2)被滿足。
還是先載入這兩個結構到VMD里并進入RMSD Calculator,左上角的文本框輸入serial 1 to 6(對應兩個分子的苯環上的碳原子序號),取消Backbone only復選框,然后點Align。按理說此時兩個分子的苯環應該被疊合了、之后再點擊RMSD就完事了,然而當前的情況比較特殊,圖形窗口里目前看到如下情況
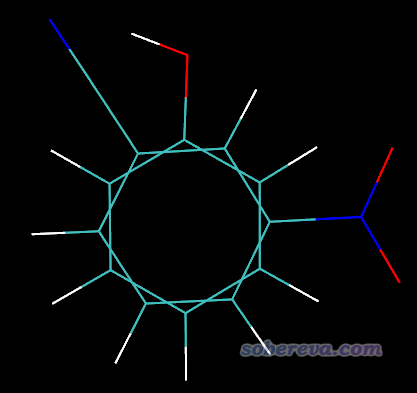
顯然還沒有真正疊合好。這是VMD用的算法的缺陷,類似于優化落到了局部極小點而非全局最小點的意味。解決辦法就是手動旋轉其中一個分子,令其苯環部分的各個原子與另一個分子的苯環部分相應序號的原子整體比較接近,類似于提供一個更好的初猜。調節結構具體的做法是選Mouse - Move - Molecule,之后按住alt鍵并用鼠標左鍵拖動某原子就可以平移整個體系,按住alt+shift并用鼠標左鍵拖動某原子可以旋轉整個分子,按住alt+shift并用鼠標右鍵拖動某原子可以令整個分子沿屏幕旋轉。等調節得差不多了,再次按Align按鈕,就會發現兩個分子的苯環部分確實很好地疊合在了一起,然后再點擊RMSD按鈕,此時情況如下所示。
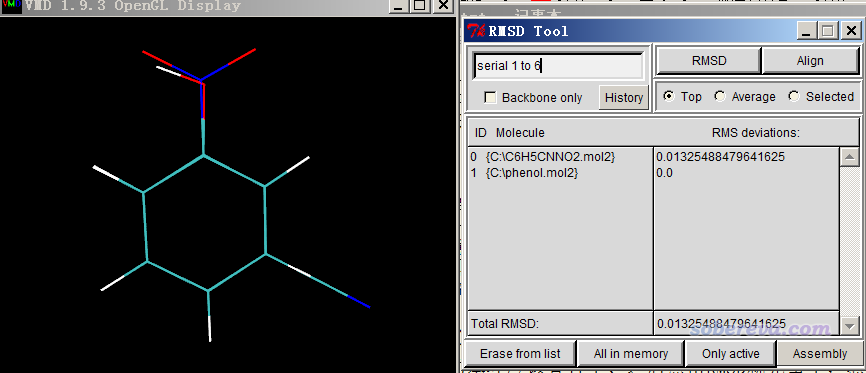
可見兩個分子的苯環碳原子間的RMSD僅為0.013埃,體現出苯環非常剛,取代基對這部分結構的影響可以忽略不計。