Yes, I did as you mentioned. I generated the surface.
Thanks
]]>"B factors in the surfanalysis.pdb are positive" is fully expected, because your system is a cation, rather than a neutral instance. For any cation, ESP on vdW surface should always be positive everywhere.
]]>Many thanks for your kind help. I adjusted the color scale in the ESPiso.vmd script in the vmd folder but again why all of the B factors in the surfaceanalysis.pdb are positive.
I didn't do the analysis by reading the .wfn file and I followed your video manual by clicking on ESPpt.bat, ESPiso.bat, and ESPext.bat, respectively to obtain the necessary files to read in vmd.
Many thanks,
Reza
]]>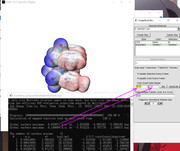
I obtained the correct ESP with MultiWFN version 2021-Sep 13 but I didn't give the same results with MultiWFN 2021, Nov. 28. I also checked the ESPiso, ESPpt, ESPext.bat.... and related.vmd files in both MultiWFN and VMD folders to be sure I copied based on the new version but again the result was a totally red colored surface.
I attach the first ESP I obtained a few weeks ago and will send you the .fchk file.
This is the pic of the ESP I obtained before:-
Regards,
Reza
If this system is a cation, you need to manually adjust color scale in "Graphics" - "Representation". Usually the lower and upper limits of the color scale can be set to minimum and maximum ESP values, respectively, then you can observe color transition on the surface. If your system is neutral, please send me your fch file and input file to me (upload to netdisk, or send to me via E-mail).
Best regards,
Tian
]]>I calculated the ESP by reading 1.fchk file of Gaussian via ESPpt.vmd, ESPiso.vmd, and ESPext.vmd routines in MultiWFN but the map is in single colour and all B factors in the Surfaceanalysis.pdb file are positive.
##################
REMARK Unit of B-factor field (i.e. ESP) is kcal/mol
HETATM 1 C MOL A 1 -4.995 -0.702 1.818 1.00 64.59 C
HETATM 2 C MOL A 1 -4.954 -0.119 -0.145 1.00 66.55 C
HETATM 3 C MOL A 1 -4.943 1.171 -1.606 1.00 65.56 C
HETATM 4 C MOL A 1 -4.370 3.483 1.896 1.00 90.91 C
HETATM 5 C MOL A 1 -3.453 -1.004 5.509 1.00 73.85 C
HETATM 6 C MOL A 1 -3.173 4.220 -3.725 1.00 76.83 C
HETATM 7 C MOL A 1 -2.859 -2.921 3.533 1.00 73.72 C
HETATM 8 C MOL A 1 -2.662 1.621 -4.339 1.00 77.09 C
HETATM 9 C MOL A 1 -2.184 0.512 5.727 1.00 72.18 C
HETATM 10 C MOL A 1 -1.858 0.190 2.412 1.00 67.20 C
HETATM 11 C MOL A 1 -1.812 2.159 -1.115 1.00 69.06 C
HETATM 12 C MOL A 1 -0.430 -2.889 -2.034 1.00 48.57 C
HETATM 13 C MOL A 1 0.445 1.946 1.923 1.00 72.20 C
HETATM 14 C MOL A 1 1.001 2.017 -2.111 1.00 70.33 C
HETATM 15 C MOL A 1 1.204 1.859 5.826 1.00 77.88 C
HETATM 16 C MOL A 1 1.448 3.427 4.917 1.00 79.99 C
HETATM 17 C MOL A 1 1.629 -3.290 -0.464 1.00 49.22 C
HETATM 18 C MOL A 1 1.764 0.330 4.609 1.00 79.73 C
HETATM 19 C MOL A 1 1.684 5.181 2.383 1.00 83.67 C
HETATM 20 C MOL A 1 1.839 5.379 0.138 1.00 92.78 C
HETATM 21 C MOL A 1 2.158 5.259 -2.063 1.00 83.23 C
HETATM 22 C MOL A 1 2.792 3.626 -4.648 1.00 78.13 C
HETATM 23 C MOL A 1 3.092 0.538 -4.402 1.00 77.11 C
HETATM 24 C MOL A 1 3.696 2.173 2.330 1.00 69.55 C
HETATM 25 C MOL A 1 4.115 1.755 0.378 1.00 70.43 C
HETATM 26 C MOL A 1 4.228 2.323 -1.586 1.00 68.66 C
HETATM 1 O MOL A 1 -6.724 -3.149 -1.899 1.00 21.96 O
HETATM 2 O MOL A 1 -5.662 0.214 2.180 1.00 57.66 O
HETATM 3 O MOL A 1 -5.622 1.654 5.921 1.00 59.35 O
HETATM 4 O MOL A 1 -5.633 1.916 -0.962 1.00 58.44 O
HETATM 5 O MOL A 1 -5.543 6.017 -1.844 1.00 60.39 O
HETATM 6 O MOL A 1 -5.140 -1.881 2.703 1.00 54.52 O
HETATM 7 O MOL A 1 -5.102 1.437 -3.040 1.00 56.01 O
HETATM 8 O MOL A 1 -1.862 1.141 2.725 1.00 63.47 O
HETATM 9 O MOL A 1 -1.851 2.914 -0.340 1.00 65.00 O
HETATM 10 O MOL A 1 -1.336 -5.830 0.171 1.00 20.41 O
HETATM 11 O MOL A 1 -1.349 -1.099 3.164 1.00 59.42 O
HETATM 12 O MOL A 1 -1.348 2.393 -2.314 1.00 62.72 O
HETATM 13 O MOL A 1 -1.257 -3.039 -4.963 1.00 25.03 O
HETATM 14 O MOL A 1 0.050 1.303 3.049 1.00 65.30 O
HETATM 15 O MOL A 1 0.402 -2.696 -0.165 1.00 39.73 O
HETATM 16 O MOL A 1 0.976 1.380 -3.483 1.00 62.21 O
HETATM 17 O MOL A 1 2.317 -4.426 2.936 1.00 23.54 O
HETATM 18 O MOL A 1 3.118 -4.192 -3.021 1.00 24.16 O
HETATM 19 O MOL A 1 3.832 2.165 3.812 1.00 58.69 O
HETATM 20 O MOL A 1 4.227 -2.764 0.636 1.00 46.56 O
HETATM 21 O MOL A 1 4.751 2.471 -3.035 1.00 57.62 O
HETATM 22 O MOL A 1 7.276 -0.857 0.830 1.00 22.79 O
END
#############################
FYI, I optimized the structure in B3LYP/LANL2DZ/6-31G* and by the optimized structure again I obtained the .fchk file with all electron basis set DZP-DKH.
Could you make a clarification.
Thanks
Reza