Please simply ignore these formal charges, they are assigned artificially and do not make great sense. You only need to properly set total net charge in the quantum chemistry calculation, only the finally obtained atomic charges can relatively faithfully represent actual charge distribution.
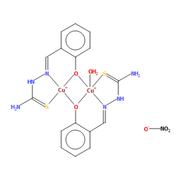
It was assigned a +2 to the di-copper-based complex and -1 to ONO2 moiety. Do I consider this situation?
]]>2 After removing the waters, I suggest performing geometry optimization for all hydrogens while keeping all heavy atoms fixed (because commonly the position of the hydrogens cannot be well determined by X-ray diffraction). Then, using the resulting wavefunction file, you can calculate atomic charges for all atoms in the coordinate as usual.
Tian
]]>I want to explore the structural properties of the copper-based complex displayed on Figure.1.
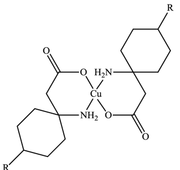
Figure.2 shows the solid-state structure of this complex (as in the CIF file): Other than the copper-based complex, of figure. 1, Figure.2 chow additional three water molecules.
The molecule in the first Figure contains 127 electrons, and that of the second figure contains 151 electrons.
My questions are:
1. Whether I used the molecule in the first figure or that of the second one.
2. There is a way to know the molecular charge in solid-state structure (as in CIF file).
Alternatively, changing, at every time, the molecular charge and optimized its geometrical parameters. The closest optimized structure to the experimental one will be taken into consideration.
thank you very much
]]>